È la famiglia di trasportatori più grande del genoma umano sono accomunati dall’ATP BINDING CASSETTE, un motivo di legame dell’ATP e da altri motivi caratteristici.
È la famiglia di trasportatori più grande del genoma umano sono accomunati dall’ATP BINDING CASSETTE, un motivo di legame dell’ATP e da altri motivi caratteristici. Nell’uomo stati individuati 48 geni appartenesti a tale superfamiglia, questi geni sono organizzati e divisi in sottofamiglie indicate con lettere che vanno dalla A alla G, di tutte le sottofamiglie, la famiglia ABCB e ABCC sono quelle che codificano per trasportatori associati a fenomeni di resistenza ai farmaci tanto che vengono anche dette MRP che sono quelle che hanno un qualche ruolo nella farmacoresistenza.
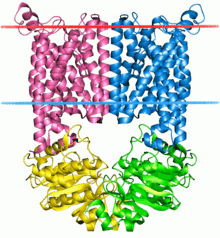
In particolare la MDR1 (multi drug resistence) o ABCB1è stata identificata negli anni ’70 in quanto era espressa da linee cellulari che mostravano una eccezionale resistenza a diversi farmaci; MDR1 è uno dei trasportatori più importanti che si caratterizza per la grandissima varietà di substrati che trasporta. Il gene che lo codifica è un gene molto lungo 209Kb e mappa sulla banda 21,5 del cromosoma 7, vi sono circa 60 SNP, la pompa di efflusso ATP dipendente ed è coinvolta nel trasporto degli xenobiotici oltre ad avere funzioni di trasporto di molecole endogene ed è espressa pressoché in tutti i tessuti dove svolge un ruolo di protezione di regolazione della componente extracellulare.
Dal punto di vista funzionale sono 2 ripetizioni di domini transmembrana e due domini per il legame di nucleotidi NB1 ed NB2 questi domini si organizzano a formare una specie di canale il cui meccanismo d’azione non è ancora chiarissimo, quello che si sa è che il legame dell’ATP determina un cambiamento conformazionale che sembrerebbe essere necessario non tanto al trasporto diretto del substrato quanto a cambiare quelli che sono o domini che interagiscono con il substrato e da una parte favorire il legame e dell’altra favorire il rilascio.
Prima di andare a vedere quali polimorfismi cercare bisogna intanto vedere se ci sono polimorfismi, le frequenze di polimorfismi sono molto diverse tra le popolazioni e la frequenza è un altro parametro importante nella scelta del polimorfismo, quando si fa uno studio di questo tipo si deve pensare alla potenza del nostro studio, se vogliamo studiare una variante particolarmente rara si deve tenere conto del numero dei soggetti che abbiamo a disposizione per studiare la variante in questione, cioè se io ho 100 persone e scelgo una variante con una frequenza inferiore all’1% al massimo se mi va bene gli alleli per quella variante saranno 1-2.
Per questo studi o polimorfismi analizzati sono C3435T e G2677A/T
– C3435T è un polimorfismo silente, si trova sull’esone 26 e comporta una sostituzione dell’allele WT G e C e negli alleli mutati diventa T (non comporta sostituzione aa);
– G2677A/T si trova nell’esone 21, l’allele WT è G mentre il mutato può essere A o T dove A è molto raro e comporta un cambiamento amminoacidico perché è un polimorfismo missenso e il cambiamento di aa è diverso a seconda che la base polimorfica sia la A (Serina) o la T (Tirosina).
Una volta scelti questi polimorfismi si passa alla fase pratica della GENOTIPIZZAZIONE e per fare questo abbiamo usato 2 metodi diversi, C3435T è stato genotipizzato con il metodo Taqman che è un metodo real time, il quale è diverso della RFLP con cui è stato genotipizzato l’altro polimorfismo.
Fondamentalmente nel metodo Taqman si usano delle sonde allele-specifiche che sono caratterizzate da un fluoroforo e un quencer. Il quencer è un silenziatore, qualcosa che impedisce al fluoroforo di emettere fluorescenza; sono delle sonde allele-specifiche, se ne usano una coppia ciascuna delle quali marcati con un fluoroforo diverso, nel caso dell’allele A per es. è marcato con un fluoroforo che si chiama BIT e nel caso dell’allele T è marcato con un fluoroforo che si chiama FAM, però quello che ci interessa è che BIT emette una fluorescenza di 510nm e FAM emette una fluorescenza di 490nm, così sono in grado di distinguerli.
L’altra cosa che ci serve per fare questi saggi è la DNA polimerasi che ha un’attività esonucleasica in direzione 5’-3’ questo perché l’appaiamento può venire in 2 maniere: quando la sonda è perfettamente complementare all’allele che c’è sul gene l’appaiamento è perfetto; quando invece la sonda non è complementare all’allele che c’e’ sul gene l’appaiamento non e’ perfetto.
Questo comporta che durante la fase di estensione la DNA polimerasi scalza la sonda senza separare il fluoroforo dal quencer e non si ha nessuna emissione di florescenza, quando invece l’appaiamento e’ perfetto diventa “PacMan” e il fluoroforo viene separato, la sonda viene degradata dall’attivita’ esonucleasica in direzione 5’-3’ dalla polimerasi il fluoroforo viene separato dal quencer e si ha emissione di florescenza che posso registrare attraverso un software tramite una curva che viene rielaborata.
Fondamentalmente si hanno dei campioni in cui soltanto uno dei reporter emette e si hanno dei campioni in cui emettono entrambi, se la sonda FAM era legata all’allele T so che quelli che emettono solo FAM avranno genotipo TT e quelli che emettono la sonda BIC saranno CC.
L’altro polimorfismo invece e’ stato genotipizzato attraverso il protocollo RFLP, la difficolta’ di questo polimorfismo e’ legato al fatto che gli alleli da discriminare sono tre e non due, e dovendo dovendone discriminare tre, si deve costruire un sistema di digestione per cui la comparazione di due digestioni diverse ci permette di determinare univocamente il genotipo.
A seconda della digestione con un enzima o con un altro abbiamo un pattern di digestione diverso, e che negli eterozigoti corrisponde a delle mutazioni diverse, comparando un pattern di digestione di un certo enzima con quello di un altro enzima si e’ in grado di determinare se questo eterozigote e’ effettivamente l’eterozigote che ha T rispetto a G (WT) .
