I target farmacologici sono localizzati in uno o più organi bersaglio, il farmaco deve arrivare al bersaglio in quantità sufficiente affinché si evochi una risposta.
Il farmaco può arrivare al bersaglio con modalità differenti; si può avere un'applicazione topica, in situ, ma spesso il target è interno e non facilmente raggiungibile, il farmaco deve essere veicolato sul bersaglio dal torrente circolatorio.
I target farmacologici sono localizzati in uno o più organi bersaglio, il farmaco deve arrivare al bersaglio in quantità sufficiente affinché si evochi una risposta.
Il farmaco può arrivare al bersaglio con modalità differenti; si può avere un'applicazione topica, in situ, ma spesso il target è interno e non facilmente raggiungibile, il farmaco deve essere veicolato sul bersaglio dal torrente circolatorio.
La concentrazione di farmaco nel sangue non è uguale a quella che arriva al bersaglio, le caratteristiche dei due sistemi sono diverse, anche se le concentrazioni sono proporzionali e variano secondo due parametri: il primo è una banale ripartizione dovuta alla lipofilicità e idrofilicità della molecola; l'altro è dovuto ad eventuali barriere fisiologiche, come la barriera emato-encefalica, in cui i capillari presentano piccolo fenestrature e sono circondati da cellule gliali rendendo il transito extravascolare più difficoltoso.
Per far entrare il farmaco si possono usare vie naturali, o enterali, e vie artificiali, o parenterali. Le vie enterali (per os, sublinguale e rettale) sono preferibili perché agevoli, indolore e non richiedono personale addetto; purtroppo sono limitate. Per le parenterali (intravascolare, intramuscolare, trans-cutanea, intratecale, intraperitoneale) si deve superare una barriera naturale. La via inalatoria è locale ma può avere un'assorbimento a livello sistemico.
Nella via intravascolare non c'è una regolazione della concentrazione dovuta all'assorbimento; in tutte le altre si deve tenere conto dell'assorbibilità sia qualitativa che quantitativa. Ciò che regola l'assorbimento è la capacità del farmaco di diffondere passivamente. Alcuni farmaci però vengono assorbiti efficacemente attraverso carrier specifici che lo velocizzano e lo rendono efficiente.
L'assorbibilità è regolata dalla legge di Fick
v = ΔC • A • K Consente di individuare le caratteristiche migliori per la
d molecola rispetto al sito di assorbimento.
Generalmente la molecola deve essere veicolata in un mezzo acquoso, ma attraversa membrane lipofile. La molecola ideale dovrebbe seguire le regole di Lipinski: affinché la molecola sia assorbibile non deve avere PM maggiore di 500; non più di 5 gruppi donatori di legami idrogeno e 10 accettori; il bilancio idrofilo/lipofilo (logP) non deve essere maggiore di 5.
se la superficie di scambio (A) è ampia la velocità di assorbimento è elevata, perciò l'assorbimento migliore si ha in distretti come intestino e alveoli polmonari. L'assorbimento è facilitato se lo spessore (d) delle membrane da attraversare è ridotto, quindi sarà più facile attraverso epiteli monostratificati. Più l'organo è irrorato più l'assorbimento è veloce.
•Via endovenosa: bypassa ogni problema di assorbimento, quantità somministrata e biodisponibile sono uguali.
•Via orale: nello stomaco l'assorbimento è limitato e trascurabile ma la presenza o meno di cibo condiziona il tempo di transito nell'intestino; anche l'acidità condiziona l'assorbimento, in teoria un acido debole dovrebbe essere maggiormente assorbibile nello stomaco, però nell'intestino si ha una maggiore area di assorbimento. Alcuni alimenti, come le mucillagini, possono impedire l'assorbimento oppure alimenti proteici possono competere con farmaci assorbiti attraverso specifici carrier. Nell'intestino si ha l'assorbimento vero e proprio, ma non è completo. Il farmaco assorbito entra nel circolo portale e passa dal fegato, dove viene in parte metabolizzato e biotrasformato (effetto di primo passaggio). La biodisponibilità orale dipende quindi da: Intestino → Circolo Portale → Fegato → Circolo Sistemico
Il metabolismo epatico influenza notevolmente la biodisponibilità. Si hanno quindi farmaci che non sono assorbiti a livello intestinale e possono essere usati a livello topico, oppure farmaci con bassa biodisponibilità perché subiscono un forte effetto di primo passaggio.
• Via sublinguale e rettale: i farmaci non passano dal circolo portale ma vengono immessi direttamente nel circolo sistemico, non vengono quindi metabolizzati dal fegato.
•Via intramuscolare: veloce, il muscolo è irrorato e i capillari presentano un'alta capacità di diffusione; la biodisponibilità è abbastanza alta. Per dilatare il tempo di azione si ricorre a farmaci in soluzione oleosa.
• Via inalatoria: elevatissima superficie di scambio e facilità di attraversamento delle membrane.
l'assorbimento, salvo casi carrier-mediati, segue una cinetica del primo ordine. La distribuzione dipende dalle concentrazioni del farmaco.
La quota di farmaco ceduta è un rapporto fisso rispetto alla quota iniziale, nell'unità di tempo.
Ct = C0 • e-kt
Cpt = C0don – (C0 • e-kt)
Dal punto di vista reale si deve tener conto anche della biodisponibilità del farmaco.
ln Ct = -kt ln Ct = -kt + ln C0 la curva è linearizzata
C0
Il farmaco ha un'emivita costante, tempo per il dimezzamento della concentrazione nel compartimento donatore: Ct = C0/2
ln Ct/C0 = -kt½
ln0,5 = -kt½
-kt½ = -0,693
kt½ = 0,693
t½ = 0,693/k
Le concentrazioni plasmatiche del farmaco sono in equilibrio con altri siti di deposito in cui viene veicolato, chiamati compartimenti di distribuzione, che possono essere acquosi o lipidici.
Il farmaco abbandona il sangue tramite un processo di eliminazione ma gli organi di deposito reimmettono il farmaco in circolo. Per farmaci in cui la distribuzione tra sangue e tessuti extravascolari è rapida si ha anche un rapido ritorno in circolo per l'eliminazione.
L'eliminazione può avvenire attraverso due modalità:
• Escrezione. Il farmaco come tale viene eliminato dall'organismo principalmente attraverso l'emuntorio renale e, in qualche caso, anche attraverso la via biliare, per farmaci lipofili. Attraverso la via biliare il farmaco può essere riassorbito nel circolo entero-epatico.
• Biotrasformazione. Il farmaco viene convertito in una molecola diversa, che può essere farmacologicamente attiva o meno.
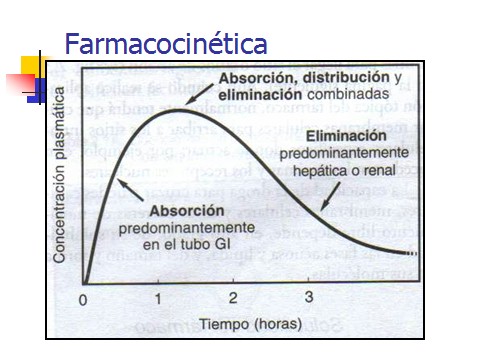
Curva dell'assorbimento di un farmaco:
Distribuzione ed eliminazione sono processi che seguono una cinetica del primo ordine. Se si somministra lo stesso farmaco alla stessa dose ma in vie diverse (per os, endovenosa) si osserva che nella curva dell'endovenosa non si ha la fase di assorbimento, il farmaco non passa da un compartimento donatore ma è già nel sangue.
Cambiando la dose varia solo Cmax e non tmax.
A parità di altre caratteristiche, più è veloce l'assorbimento più si raggiunge brevemente la concentrazione plasmatica.
Il farmaco, per essere efficace, deve raggiungere una certa concentrazione plasmatica; infatti l'effetto a livello del sito bersaglio è in funzione della concentrazione.
Aumentando la dose aumenta il tempo di efficacia. Esiste però anche una concentrazione tossica, che non deve essere raggiunta.
Se l'indice terapeutico del farmaco è piccolo deve essere considerata la via di somministrazione, una via più rapida porta ad un più rapido assorbimento ma anche ad un picco più alto.
A parità di dosaggio e farmaco si ha l'area più grande sotto la curva (AUC) per la via endovenosa. La curva di questa via, non presentando assorbimento, è determinata solo dall'eliminazione, la stessa per tutte le vie di somministrazione; la differenza di AUC tra la via endovenosa e le altre è espressione della biodisponibilità, quindi del mancato assorbimento o dell'assorbimento ridotto dovuto alla metabolizzazione del farmaco.
Biodisponibilità = AUCx/AUCen
La maggior parte dei farmaci ha una farmacocinetica lineare, in rari casi alcuni hanno un andamento che non può essere descritto da regole matematiche, quindi sono difficilmente gestibili.
La distribuzione avviene in compartimenti extravascolari ma nel sangue esistono delle proteine plasmatiche (albumina, α1-antitripsina) con particolare affinità per i farmaci. Il farmaco segue quantitativamente le stesse regole della formazione del legame farmaco-recettore. Questo legame esiste a livelli elevati per farmaci con scarsa solubilità in acqua. Solitamente una proteina plasmatica può avere più siti di legame con il farmaco; per la sua funzione finalistica la proteina è fatta per legare tutte quelle sostanze insolubili in acqua, quindi il legame è aspecifico.
Il farmaco legato alla proteina non può essere filtrato a livello renale e non lascia il torrente circolatorio, perciò gli effetti di questo tipo di farmaci sono regolati dalla concentrazione plasmatica di farmaco libero.
F + P ↔FP
Ftot = Fb + Ff
Se FP < 20% non si hanno complicazioni.
Se 20% < FP < 90% il farmaco deve essere gestito caso per caso.
Se FP > 90% il farmaco è fortemente legato e va gestito con molta attenzione.
All'effetto contribuisce solo Ffree; si deve quindi stare attenti all'interazione causata da altri farmaci affini alle proteine plasmatiche che possono spostare l'equilibrio causando uno sbalzo nelle concentrazioni plasmatiche che può portare, in caso di IT basso, ad effetti tossici.
La concentrazione teorica (senza tener conto dell'eliminazione) di farmaco nel sangue non è data solo dalla dose nel volume plasmatico (0,07 l/kg), ma tiene conto anche della distribuzione nei compartimenti acquosi (0,7 l/kg).
Cp = D / Vd Vd = D / Cp
Quindi data una determinata dose di farmaco, per esempio 1g, e sapendo che nel sangue si ha il 10% della dose (0,02g/l) si può calcolare il volume apparente di distribuzione che è Vd=1/0.02=50l
Se Vd ~ 5l il farmaco è per la maggior parte distribuito nel sangue, legato alle proteine plasmatiche;
se Vd ~ 50l il farmaco è idrofilo e distribuito nei compartimenti extracellulari; se Vd ~ 500l il farmaco si distribuisce in tessuti quali l'adiposo, l'encefalico e quello muscolare.
Vd non è il volume ematico vero e proprio ma il volume ematico calcolato come se il farmaco fosse solo nel sangue.
Dal punto di vista pratico la Cmax è maggiore rispetto a quella teorizzata, il farmaco assunto deve raggiungere l'equilibrio.
Ct = Co (1-k)t Co = D / Vd
Ct = D / Vd (1-k)t
La Co calcolata non considera la fase di distribuzione, ma solo quella di eliminazione. Lo stesso concetto si applica al farmaco somministrato per vie diverse da quella endovenosa.
A parità di farmaco e dose si ha la stessa k per vie di somministrazione diverse. La k corrisponde alla quota di farmaco eliminato per unità di tempo.
Cl = Velim /t = 0,693 /t½
L'unico fattore che influenza le aree
dei farmaci è la cinetica di eliminazione.
Cl = D / AUC
Questo parametro è fondamentale per
regimi di somministrazione continuata.
Cl • Cp = Felim /t = υelim
Per mantenere fissa la concentrazione plasmatica si deve avere υelim = υsomm
Data una certa biodisponibilità: F = Dos / Den = AUCos / AUCen
Si può tranquillamente ricavare che: Clos = Clen
Clos = Dos / AUCos
Dos = F • Den
AUCos = F • AUCen
Clos = F • Den = Clen
F • AUCen
Il farmaco in endovena viene somministrato per diffusione continua; se si effettuano somministrazioni ad intervalli regolari la velocità teorica rimane la stessa ma in realtà si hanno oscillazioni della velocità reale. Aumentando il tempo tra una somministrazione e l'altra aumenta l'ampiezza dell'oscillazione. Cpi reale è data dalla somma dei contributi delle singole somministrazioni.
Quanto tempo serve per raggiungere lo stato stazionario? Supponiamo di somministrare un'unità posologica ogni emivita. Cp = 100
Prima emivita: 50 + 100 = 150
Seconda emivita: 75 + 100 = 175
Terza emivita: 87,5 + 100 = 187,5
Quarta emivita: 93,75 + 100 = 193,75
Quinta emivita: 96,875 + 100 = 196,875
Lo stato stazionario si raggiunge in circa 4/5 emivite, qualunque sia la velocità di somministrazione.
Raddoppiando la dose il tempo di eliminazione aumenta di un'emivita.
Un farmaco con un'emivita molto lunga, che necessita di molte ore perché si osservi un effetto, ha bisogno di una prima somministrazione con una dose di carico Dcarico = C • Vd
La clearance totale è dovuta quasi esclusivamente alla clearance renale, anche se è presente una clearance non renale (solitamente epatica).
Il farmaco, se sufficientemente piccolo, a livello glomerulare viene completamente filtrato. Nel nefrone può eventualmente tornare nel corpo, dipende dalle caratteristiche di farmaco e membrana: se idrofilo o carico viene filtrato, se presenta buone caratteristiche di bilancio idrofilo/lipofilo ha un'ottima capacità di essere riassorbito, fino al raggiungimento dell'equilibrio tra [Ff] e [Fint]; il farmaco filtrato e non riassorbito viene concentrato ed eliminato con le urine.
Esempio. Un farmaco con un'ottima densità di carica e idrofilicità viene somministrato per via parenterale. Qual'è il suo volume di distribuzione? Dipende solo da plasma e liquido extracellulare, non diffonde nel citosol a meno che non ci siano meccanismi di trasporto attivo.
Molecole dissociate presentano una clearance alta perché filtrate a livello glomerulare, ma non soggette a riassorbimento a livello tubulare. In questi casi la clearance tende a sovrapporsi a quella che è l'entità della filtrazione. Quando invece ci si allontana da questo estremo e la molecola ha caratteristiche di maggiore riassorbibilità, viene filtrata a livello del glomerulo ma nei tubuli in parte viene riassorbita, fino all'eccesso opposto ossia che il riassorbimento sia tale per cui la concentrazione del filtrato ritorni a essere uguale alla concentrazione all'interno dell'organismo.
Esempio. Si tiene conto solo della caratteristica chimico fisica relativa al riassorbimento. Nel primo caso quello che viene filtrato è uguale a quello che sparisce. In un altro caso in parte viene richiamato mettendosi in equilibrio, insieme all'acqua. Quindi, qual'è il volume di questo farmaco, che è uguale alla concentrazione che in quel momento è nell'organismo, nel torrente circolatorio, viene buttato fuori?
In questo caso la clearance sarà di 1,5 litri al giorno (pari alle urine eliminate). Siamo sugli 0,6 litri l'ora.
