- Il diabete di tipo 1, detto anche insulino-dipendente, in genere insorge nell’infanzia o nell’adolescenza.
- Il diabete di tipo 2 è la forma più comune e rappresenta il 90% circa dei casi oggi diagnosticati.

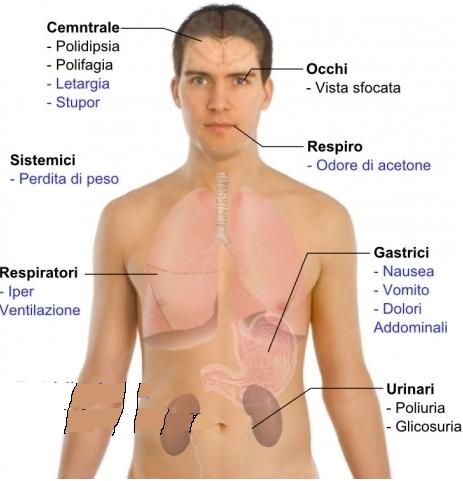
Si tratta di un disordine cronico del metabolismo dei carboidrati, lipidi e proteine, causato da una risposta secretoria insufficiente o difettosa dell’insulina, che determina una compromissione del metabolismo dei carboidrati (glucosio), con conseguente iperglicemia.
Classificazione: le forme più importanti di diabete mellito originano da disordini primitivi del sistema di segnale insulina-cellula insulare.
DIABETE di TIPO 1 o DIABETE MELLITO INSULINO DIPENDENTE (IDDM) (diabete ad insorgenza giovanile) (10%)
DIABETE di TIPO 2 o DIABETE MELLITO NON INSULINO DIPENDENTE (NIDDM) (80-90%)
DIABETE GIOVANILE AD INSORGENZA IN ETA’ ADULTA (MODY) → difetti genetici della funzione delle cellule β (malattia autosomica dominante) (5%)
Mentre i 2 tipi principali di diabete presentano caratteristiche metaboliche e meccanismi patogenetici diversi, le complicanze croniche a lungo termine a carico dei vasi, dei reni, degli occhi e dei nervi sono comuni ad entrambi i tipi, costituendo le cause principali di morbidità e mortalità nella malattia.
Rappresenta l’espressione clinica di un gruppo eterogeneo di lesioni genetiche della funzione delle cellule β caratterizzato da:
ereditarietà autosomica dominante di un difetto monogenico ad alta penetranza;
insorgenza precoce, in genere prima dei 25 anni, rispetto ai pazienti con diabete di tipo 2;
compromissione della funzione delle cellule β, peso normale, assenza di Ab GAD ed assenza della sindrome da resistenza insulinica.
4 difetti genetici:
MODY1: mutazione del gene per il fattore di trascrizione nucleare epatocitario 4a (HNF-4a), posto sul cromosoma 20, responsabile della regolazione dell’espressione di HNF-1α;
MODY2: difetto al gene della glucochinasi sul cromosoma 7;
MODY3: mutazioni a carico del gene per il fattore epatocitario nucleare 1α (HNF-1α), posto su 12q. L’HNF-1α è un fattore di trascrizione che funziona come transattivatore del gene dell’insulina-I;
Mutazioni puntiformi del DNA mitocondriale.
Suscettibilità genetica: almeno uno dei geni della suscettibilità si trova nella regione del cromosoma 6p21, che codifica per gli Ag di classe II del sistema maggiore di istocompatibilità (HLA-D). HLA-D → 3 classi di geni –DP, DQ e DR, che contengono, ciascuno numerosi alleli.
Il 95% dei pazienti bianchi affetti da diabete mellito di tipo 1 presentano l’allele HLA-DR3 o l’allele HLA-DR4 o entrambi. Associazione ancora più forte con alcuni alleli nel locus DQ, che si trovano in “linkage disequilibrium” (ereditati insieme) con i geni HLA-DR.
Autoimmunità: questa malattia è espressione di un attacco autoimmune cronico alle cellule β (presente anni prima che si manifesti la malattia).
Un difetto primario dell’immunità può dar luogo a molecole aberranti di classe II nelle cellule β. Ciò porterebbe al riconoscimento di Ag propri di tali cellule da parte dei linfocit T-helper con conseguente infiltrazione linfocitaria, inducendo, quindi, la liberazione di citochine INFγ nell’infiltrato, con ↑ dell’espressione degli Ag di classe I.
Fattori ambientali: possono innescare l’autoimmunità. Un possibile ruolo potrebbe essere svolto dai VIRUS (Coxsachie B, morbillo, parotite, citomegalovirus, rosalia e mononucleosi). Varie ipotesi: il diabete di tipo 1 potrebbe essere una rara conseguenza di alcune infezioni virali comuni, ritardato dal lungo periodo di latenza necessario perché si realizzi la perdita immunomediata di cellule β e dipendenti dagli effetti modificatori del background genetico; la risposta immune potrebbe svilupparsi contro una proteina virale che condivide delle sequenze aminoacidiche con una proteina delle cellule β (“mimetismo molecolare”).
Altro: derivati del latte vaccino; tossine chimiche.

Le disfunzioni del lobo posteriore ipofisario sono rare e per lo più correlate alle lesioni sopra-sellari ipotalamiche. Le alterazioni del lobo posteriore si manifestano essenzialmente con l' insufficiente secrezione di ADH.
La deficienza di ADH causa il diabete insipido, caratterizzato da poliuria, sete eccessiva e polidipsia.
Le cause di questa sindrome possono essere:
1) alterazioni infiammatorie o neoplastiche dell' asse ipotalamo – ipofisario (come tumori sopra-sellari, metastasi, ascessi, meningiti, tubercolosi o sarcoidosi);
2) danni chirurgici o da radioterapia cioé ipofi-sectomia chirurgica o da raggi;
3) severi traumi cranici;
4) cause idiopatiche.
L' inappropriata secrezione di ADH implica una secrezione ormonale svincolata dai valori di osmolarità del plasma. Quindi si ha eccessivo riassorbimento di acqua dal filtrato glomerulare, ritenzione idrica con espansione del volume extracellulare, con conseguente iponatriemia ed emodiluizione ed inabilità a produrre urine diluite.
La causa più comune é la secrezione di ADH da parte di varie neoplasie (sindrome paraneoplastica); in particolare il carcinoma a piccole cellule del polmone costituisce la causa di oltre i quattro quinti dei casi. Più raramente alterazioni del sistema nervoso centrale, quali emorragie intracerebrali o sub-aracnoidee, trombosi, ematomi sub-durali o infezioni possono produrre la sindrome, cosí come le malattie polmonari, quali polmonite e tubercolosi ed infine alcuni farmaci.
E’ un disordine complesso e multifattoriale, coinvolgente sia l’alterato rilascio di insulina, che conduce ad un deficit relativo di insulina, sia l’insensibilità degli organi periferici a rispondere alla secrezione insulinica (insulino-resistenza).
Non esiste correlazione con il sistema HLA.
Disregolazione della secrezione di insulina:
perdita della secrezione pulsatile di insulina
perdita della fase precoce della risposta di insulina
Resistenza insulinica: può essere dovuta ad una diminuzione del numero dei recettori o ad un difetto post-recettoriale (es.ridotta sintesi e/o traslocazione di GLUT nel muscolo e negli adipociti).
L’insulino resitenza, indipendentemente dai meccanismi che la generano, determina:
incapacità dell’insulina circolante di modulare la disponibilità di glucosio
iperglicemia persistente
prolungata stimolazione delle cellule β
Amilina: peptide prodotto normalmente dalle cellule, impacchettato con l’insulina e cosecreto negli spazi sinusoidali. Nei pazienti con diabete mellito tipo 2, l’amilina tende ad accumularsi al di fuori delle cellule β, in stretto contatto con la membrana cellulare (amiloide).