Talassemia α: α+: ridotta sintesi delle catena α dell’emoglobina;
α0: assente sintesi della catena α dell’emoglobina;
Talassemia β: β +: ridotta sintesi delle catena β dell’emoglobina;
Talassemia α: α+: ridotta sintesi delle catena α dell’emoglobina;
α0: assente sintesi della catena α dell’emoglobina;
Talassemia β: β +: ridotta sintesi delle catena β dell’emoglobina;
β0: assente sintesi della catena β dell’emoglobina;
Su scala mondiale le talassemie sono le più frequenti malattie geneticamente determinate, frequente nei paesi mediterranei nonché in altri paesi tra cui quelli in cui la malaria è endemica.
La malattia porta sia ad un basso livello di emogobina ma anche ad un relativo eccesso di una delle due catene.
Talassemie α
Dal punto di vista dei difetti molecolari nella maggior parte dei casi il problema è la delezione di uno o più de i 4 geni (2 per ogni cromosoma 16, ognuno che contribuisce per il 25%) che codificano per la globina α.
Esistono anche mutazioni più rare, che non sono delezioni:
o Mutazioni del codone d’inizio o della sequenza immediatamente a monte;
o Mutazioni non senso che inseriscono un codone di STOP;
o Mutazioni nel sito di poliadenilazione.
Per quanto riguarda le delezioni, esse avvengono solitamente per crossing over ineguale poiché i due geni ripetuti possono, nel corso della meiosi, appaiarsi in maniera diseguale. Si ottiene così un cromosoma con tre geni ed uno con un solo gene.
Più geni sono presenti, meno grave è la malattia. Difatti:
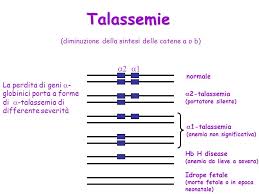
o Tre geni (eterozigote αα/α-): l’individuo è portatore silente, asintomatico e privo di anomalie strutturali nel sangue;
o Due geni (–/αα oppure –α/-α): asintomatico ma con lieve anemia emolitica e con alcune cellule microcitiche. Si parla di “tratto α-talassemico”.
o Un gene (–/-α): moderata anemia emolitica con ipocromia e microcitosi. Il problema maggiore è in questo caso l’eccesso di catene β: si forma difatti la cosiddetta emoglobina H, formata da quattro catene β, che per le sue caratteristiche precipita nei globuli rossi diminuendone l’emivita e accelerandone l’emolisi. HbH ha inoltre alta affinità per l’ossigeno;
o Assenza di geni: la condizione è detta idrope fetale e non è compatibile con lo sviluppo intrauterino del feto in quanto si formano tetrameri di catene γ (Hb di Bart) la quale ha altissima affinità per l’ossigeno.
Talassemie β
A livello molecolare questa malattia è raramente causata da delezioni mentre molto più frequenti sono le mutazioni puntiformi. In particolare quest’ultimo tipo di mutazione possono modificare:
o La trascrizione (mutazione del TATA box oppure in una sequenza importante per la trascrizione situata 90 basi a monte del sito d’inizio); β+
o Il significato dell’RNA: mutazioni non senso o frameshift; β0
o Lo splicing:
o Mutazione di una base nel sito di splicing nella giunzione tra esone ed introne. La mutazione annulla completamente la sintesi della globina; β0
o Creazione di un sito di splicing alternativo; β+
o Mutazione nei siti di poli adenilazione. β+
Anche se rare vanno comunque citate le delezioni. La più frequente di esse è dovuta ad un appaiamento anomalo nella meiosi tra i geni per le globine δ e β e formazione di un gene di fusione δ/β (Hb lepore). Il promotore del gene della globina δ è molto debole e l’espressione è bassa.
Le conseguenze dell’alterata sintesi delle catene β sono:
o Insufficiente produzione di HbA con globuli rossi microcitici ed ipocromici;
o Squilibrio tra la sintesi delle globine α e β: le catene α in eccesso si aggregano e precipitano nei globuli rossi determinandone la morte per apoptosi già allo stadio di eritroblasti (eritropoiesi inefficace). Gli eritrociti che sopravvivono vengono comunque eliminati nella milza;
Anche nel caso delle talassemie β, il quadro clinico dipende da quanti e come i geni sono colpiti. Si distinguono:
o Talassemia maior (omozigosi β0/ β0 o β+/ β+): provoca anemia microcitica ipocromica, emolisi gravi, epatosplenomegalia, iperplasia midollare (a causa dell’aumentata eritropoiesi) che provoca deformità dello scheletro. A causa delle ripetute trasfusioni si determina sovraccarico di ferro, che è la causa maggiore di danno soprattutto a carico di reni e milza. La mancanza di catene β non è comunque incompatibile con la vita.
o Talassemia minor (eterozigosi β0/ β o β+/ β): si verifica modesta riduzione di HbA, aumento di HbA2 (α2δ2), lieve anemia con ipocromie.
