Cosa significa Trasduzione del Segnale in patologia generale
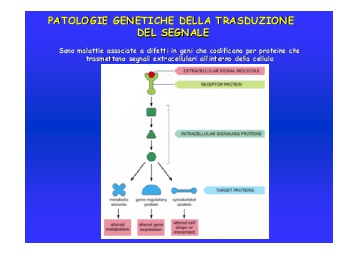
Sono malattie associate a difetti in geni che codificano proteine che trasmettono segnali dall’esterno all’interno delle cellule. Ad ogni ligando corrisponde un recettore. Per esempio i recettori per fattori di crescita sono tirosin chinasi. Altri tipici recettori sono quelli a 7 domini transmembrana (es. recettori per ormoni), altri ancora quando legano il ligando reclutano una tirosin chinasi citosolica (es. recettori per citochine).

Le proteine di trasduzione, attivate dai recettori, innescano una serie di reazioni a cascata che portano per esempio ad attivazione dei fattori di trascrizione. Numerose malattie genetiche sono dovute a difetti nelle vie di trasduzione del segnale.
Vie dei fattori di crescita
I recettori sono delle tirosin chinasi i cui domini extracitoplasmatici possono essere di diverso tipo (domini ricchi di cisteine, domini Ig-simili…). In seguito al legame del fattore di crescita epiteliale (EGF) al recettore questo dimerizza e ciascuna subunità fosforila l’altra. In seguito all’autofosforilazione si ha reclutamento di proteine adattatrici. Una di esse è GRR2, che ha due domini: uno SM2 che interagisce coi siti fosforilati e un altro, SM3, che lega SOS. La proteina SOS a sua volta lega RAS.
La proteina SOS
La SOS fa parte della famiglia GEF (GDP exchanging factor). Essa fa sì che RAS, normalmente inattiva e legante GDP, si attivi liberando il GDP e legando GTP. Questo è un processo fondamentale per la successiva trasmissione del segnale. Difatti RAS attiva RAF, una proteina che fa parte delle MAP-chinasi (attivatore delle capacità mitogeniche). Le MAP-chinasi sono enzimi in grado di fosforilare residui di serina/tiroxina. Le MAP-chinasi sono tre ed ognuna fosforila la successiva. L’ultima trasloca nel nucleo ed induce l’attivazione di fattori di trascrizione con sintesi di proteine che portano alla progressione del ciclo cellulare e l’entrata della cellula in mitosi.
Nella cellula la disattivazione di RAS è effettuata ad opera della famiglia delle proteine citosoliche GAP (GTPase activating protein) che amplificano la debole attività GTPasica di RAS.Sono state identificate numerose mutazioni che portano ad una attivazione costitutiva di RAS, la quale comporta proliferazione neoplastica. Si può avere proliferazione neoplastica anche per difetti genici di GAP, i quali determinano la mancata inattivazione di RAS.
Neurofibromatosi
La neurofibromatosi di tipo I, è una malattia AD con un’incidenza di 1/3500 tanto da essere considerata la più frequente malattia neurologica genetica. E’ una malattia a penetranza completa ma ad espressione variabile, 1/3 dei casi deriva da nuove mutazioni.
Il gene NF1 è localizzato nel cromosoma 17 e codifica per la neurofibrosina. Essa è trascritta in tutte le cellule ma l’espressione avviene soprattutto nel SNC e nelle cellule cromaffini del surrene.
Il risultato finale della mutazione è una perdita di funzione. L’inattivazione della proteina porta all’alterazione delle vie di trasmissione del segnale regolata dalle G proteine “RAS like”. Difatti la neurofibrosina non è altro che un tipo di proteina GAP e può essere classificata come un oncosoppressore essendo essa un interruttore molecolare che spegne RAS. In assenza della proteina si assiste ad una proliferazione cellulare sregolata.
Questa malattia genetica è caratterizzata dalla comparsa di vari tumori, soprattutto originati da cellule del neuroectoderma:
- Neurofibromi (tumori benigni dei nervi periferici dovuti a proliferazione sregolata delle cellule di Schwann e dei fibroblasti perineuronali ed endoneuronali);
- Amartomi dell’iride;
- Macchie cutanee (macchie dette a “caffè-latte” per il loro colore);
- Ritardo mentale (non sempre e non è comunque un segno patognomico);
- Aumentato rischio di insorgenza di alcuni tumori maligni (neurofibrosarcomi, astrocitomi, rabdomiosarcomi).
La sintomatologia insorge in età adolescenziale o anche più tardi.
Retinite Pigmentosa
Con il termine di retinite pigmentosa (RP) si descrive un gruppo di malattie in quanto più difetti genici possono portare allo sviluppo dello stesso fenotipo anche se il gruppo è geneticamente e clinicamente eterogeneo. I difetti genici sono tutti a carico di proteine della via di trasduzione del segnale e possono essere AD, AR e legate all’X.
Le malattie sono caratterizzate da degenerazione retinica con perdita di recettori visivi con proliferazioni focali dell’epitelio retinico pigmentato. La rodopsina ha la struttura tipica dei recettori a 7 domini transmembrana ed è legata ad una proteina G trimerica. Sui dischi dei bastoncelli ci sono numerosi canali per il sodio cGMP dipendenti. La membrana quindi, normalmente, è depolarizzata.
I recettori a 7 domini transmembrana sono sempre associati a proteine G trimeriche. In assenza di segnale la proteina G lega il GDP. Quando il recettore si lega al ligando avviene un’attivazione della proteina G che rilascia il GDP e lega il GTP. Questo fa sì che la subunità α si stacchi da βγ e vada ad agire a livello di un effettore, diverso a seconda della via di trasduzione. Nel caso della via di traduzione del segnale luminoso il ligando è un fotone, il recettore è la rodopsina e la proteina G trimerica è detta transducina (GT). L’effettore che viene attivato in seguito al legame con la subunità Gα è la fosfodiesterasi. Ciò determina la chiusura del canale del sodio e una conseguente iperpolarizzazione.
Alcune forme di RP autosomiche dominanti sono dovute ad un difetto nel gene della rodopsina nel cromosoma 3. Nell’80% dei casi si tratta di mutazioni puntiformi. La rodopsina mutata non viene trasportata nella membrana dopo la traduzione. Alcune forme mutate vengono trasportate ma destabilizzano la membrana portando così a morte i bastoncelli: ciò causa deposizione di pigmento e perdita di visibilità notturna.
La morte dei bastoncelli può provocare degenerazione della retina con coinvolgimento anche dei coni e perdita anche della visibilità diurna. Altri casi derivano da mutazioni della fosfodiesterasi o dei canali del sodio.
