In queste malformazioni c’è una precoce fusione che porta all’immobilazione di crescita di quella parte o di quelle parti del cranio e della faccia, infatti si parla di cranio-facio-stenosi, andiamo sempre all’embriologia.
Ricordiamoci le suture craniche e le famose fontanelle nel neonato dove abbiamo:
- una sutura sagittale,
- una sutura lambdoidea,
- una sutura coronale.
Se il parietale con il suo omonimo controlaterale si ossifica prima delle altre ossa, essendo solidarizzate queste ossa mentre il resto si evolve naturalmente vi dovreste aspettare un allungamento antero-posteriore.
Distinguiamo forme sindromiche e non sindromiche
Craniostenosi primarie
Anche dette semplici, sono le più frequenti.
Comprendono deformazioni:
- forme monosuturali
- la scafocefalia,
- la plagiocefalia,
- la trigonocefalia;
- forme plurisuturali:
-
- brachicefalia,
- oxicefalia.
Craniostenosi sindromiche
Nelle forme sindromiche (anche dette complesse) abbiamo:
- sindrome di Crouzon,
- sindrome di Apert,
- sindrome di Pfeiffer,
- sindrome di Nuenke
- sindrome di Cothzen-Sarthre.
Le suture
Le suture sono:
- la coronale,
- la sagittale,
- la lamboidea,
- la metopica (o frontale) ed altre.
La metopica non si riscontra come sutura vera e propria perchè si fondono tra di loro poco intensamente e molto rapidamente è quindi una sutura temporanea; compensa lo sviluppo molto rapido dell’encefalo nella vita pre e post natale.
L’ultima fontanella a chiudersi cronologicamente è la bregmatica.
La Crouzon e l’Apert sono le più frequenti e sono similari.
Uno dei migliori infanticidi era di mettere uno spillone nella fontanella bregmatica, cosi facendo si procedeva facilmente all’aggressione cefalica del neonato.
Le fontanelle
- La prima fontanella è la bregmatica,
- la seconda la lamboidea,
- l’ultimo punto a chiudersi,
- l’altra fontanella è la pteriodea.
Craniostenosi non sindromiche dette anche semplici o primarie
Craniosinostosi non sindromiche:
- Scafocefalia: 25% di tutte le craniosinostosi, fusione precoce sutura sagittale e cranio allungato antero posteriormente e ristretto in senso latero-laterale, bozze frontali prominenti.
- Brachicefalia: fusione coronale, cranio appiattito. Diametro antero posteriore diminuito, compenso verticale, quindi un cranio corto ma largo ed elevato.
- Plagiocefalia: anteriore e posteriore. L’anteriore è il 12% di tutte le craniosinostosi, fusione precoce della emicoronale; alterazioni patologiche craniche e facciali perchè si associa a fusione di alcune sincontrosi, cranio appiattito. [plagiocefalia falsa da posizionamento] Esiste la forma posteriore molto più rara.
- Trigonocefalia: regione frontale sollevata, cresta mediana che va dalla regione bregmatica alla glabella,bozze frontali non rappresentate.
Oxicefalia: consiste nella stenia di più suture craniche.
Molto raro è il cranio a trifoglio o “Kleeblattschädel”.
Nella pansinostosi tutte le suture si chiudono prima ed abbiamo la microcefalia.
Nella trigonocefalia si chiude prima la metopica.
In corrispondenza della prematura fusione della sutura cranica si osserva l’arresto della crescita del cranio nella direzione perpendicolare e lo sviluppo compensativo del cranio principalmente secondo la direzione della sutura stessa.
Sindrome di Couzon
Patologia in questo caso autosomico dominante per un recettore, FGFR2 (recettore del fattore di crescita del fibroblasto 2) , conformazione cranica brachicefalica o oxicefalica.
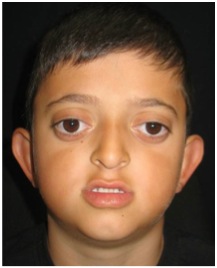
Si mostra ipertelorismo esoftalmo, ipoplasia facciale, sindattilia.
C’è un iposviluppo del terzo medio del volto, lo sfenoide è sviluppato con alterazione della grande ala che produce una diminuizione della profondità dell’orbita, l’occhio essendo normale protude troppo. Quindi è la base cranica ad essere coinvolta.
Sindrome di Apert o acrocefalosindattilia ereditaria
Simile alla sindrome di Crouzon, è lo stesso momento patogenetico ma c’è qualcosa in più. Guardate mani e piedi, se c’è qualche fusione delle dita oltre all’iposviluppo del volto come la sindrome di Crouzon siete davanti ad una sindrome di Apert. È caratterizzato clinicamente da:
- brachicefalia,
- esoftalmo asimmetrico associato a ptosi palpebrale
- sindattilia anche totale (mano a cucchiaio).
Più frequente l’idrocefalo.
Autosomica dominante con un’alterazione dello stesso gene della Crouzon.
