F + R ↔ FR ~ effetto (visibile a vari livelli)
La reazione è regolata da una costante di dissociazione (Kd), all’equilibrio si ha quindi una determinata quantità di complesso che è direttamente proporzionale alla [F] presente.
Esiste una [F] soglia, al di sopra della quale è inutile aumentare l’agonista (in questo tipo di sistema) perché non si ha più aumento di [FR].
La curva è definibile anche come FR = Rt
1 + Kd/F
Se F = Kd allora FR/Rt= ½ in questo caso si satura la metà dei recettori totali disponibili.
Devono essere presenti zone di complementarietà chimica tra il farmaco e il recettore, più sono più il farmaco è affine al recettore e più è improbabile che abbia affinità per altri recettori. Nell’interazione con recettori simili l’equazione rimane invariata, l’unico parametro che varia è la Kd.
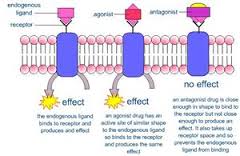
Farmaco agonista
Un farmaco agonista somministrato produce un’azione sul soggetto di studio dose-dipendente. La curva che si ricava, anche in questo caso, ha un andamento di tipo sigmoidale, come quella che correla il farmaco al recettore. L’effetto massimale è il maggiore effetto funzionale possibile prodotto da quel dato agonista in quel dato sistema.
Eff = 1
Emax 1 + X/F
X è la concentrazione tale per cui quando
X = F si ha Eff = Emax/2
Il nome corretto di questo parametro è EC50 per sistemi in vitro e ED50 per sistemi in vivo.
La teoria di Clark
Secondo Clark la risposta osservata è funzione diretta dell’occupazione recettoriale; quindi l’equazione si può scrivere come
Eff = x . FR
Emax Rt
quindi [F] in grado di produrre la metà dell’efficacia totale è uguale a [F] in grado di occupare la metà dei recettori liberi.
Secondo questa teoria le due curve dovrebbero essere indistinguibili; Clark però non sapeva che quando si innesca un complesso farmaco-recettore esso, a sua volta, innesca una serie di reazioni biochimiche ognuna con un proprio equilibrio e una diversa Kd (per le interazioni chimiche reversibili).
Se i processi a valle sono più favoriti si ha un sistema amplificato, quindi la curva dell’efficacia è spostata a sinistra rispetto a quella dell’occupazione recettoriale, l’effetto massimale quindi non è dato dalla massima occupazione recettoriale ma dall’esaurimento di sistemi a valle. Il numero di recettori è pleonastico, i recettori in eccesso sono chiamati riserva recettoriale, il loro significato biologico varia da sistema a sistema.
La funzione può essere scritta come Eff/Emax = ƒ(FR/Rt)
Se viene utilizzato un diverso farmaco sullo stesso recettore si ha un effetto concentrazione-dipendente per cui si raggiunge la stessa Emax con una diversa EC50. Il parametro di EC50 in una curva graduale determina la potenza di un farmaco. L’efficacia o attività intrinseca è il rapporto tra l’effetto massimo del farmaco e l’effetto massimo totale possibile in quel sistema per quel dato recettore. Se l’efficacia possibile e quella totale coincidono il farmaco è un agonista puro, in caso contrario è un agonista parziale.
Per un antagonista la curva di occupazione recettoriale è uguale a quella di un agonista, ma la curva dell’effetto è nulla, non si hanno infatti effetti visibili. L’unico modo per studiare la sua efficacia è attraverso una curva concentrazione-risposta di un agonista in presenza dell’antagonista in esame.
L’equazione di Hill completa è Eff = 1 generalmente n tende a 1
Emax 1 + (EC50/F)n
Un agonista parziale con un’efficacia vicina allo zero è molto più simile a un antagonista perché impedisce il legame della molecola intrinseca al suo recettore.
Gli antagonisti recettoriali impediscono che l’agonista si leghi al recettore; da un punto di vista funzionale esistono altri tipi di antagonisti, come acetilcolina e noradrenalina.
In assenza di antagonista l’agonista interagisce con la popolazione recettoriale formando un complesso che possiede una data attività:
A+R↔AR con attività ƒ(AR)
In presenza di antagonista si instaurano legami col recettore analoghi a quelli dell’agonista formando un complesso BR inattivo.
B In questo caso [AR] dipende da [B] e dalla Kb; un
+ Ka antagonista di questo tipo si chiama competitivo
A + R ↔ AR reversibile o sormontabile.
↕Kb In questo caso si osserva uno spostamento parallelo delle
BR curve concentrazione-risposta; l’ampiezza è proporzionale alla concentrazione e alla K dell’antagonista.
Indice di potenza dell’antagonista
Eff= 1 Eff = 1
Emax 1+(EC50/A) Emax 1+(EC50‘/A)
EC50‘ = EC50 (1+B/Kb)
In questa famiglia di curve esistono concentrazioni equiattive, che possono essere chiamate A e A’.
Quindi si ha:
1 1
1 + EC50 1 + EC50(1+B/Kb)
A A’
1 + EC50 1 + EC50 (1+B/Kb)
A A’
EC50 EC50 (1+B/Kb)
A A’
A’=A(1+B/Kb)
A’= 1 + B B = A’ – 1
A Kb Kb A
DR – 1 = B/Kb
Da ciò si ricava l’equazione di Schild.
Equazione di Schild
Log(DR-1) = LogB – LogKb
Questa equazione in forma logaritmica rappresenta una retta. Se la pendenza della retta è pari a 1 allora lo Schild plot è diagnostico di un antagonista reversibile.
L’antagonista può essere utilizzato anche per ridimensionare un sito recettoriale sovrastimolato; viene usato, per esempio, per intossicazioni da organofosforici o gas nervini, che portano ad un incremento di acetilcolina (uso di atropina); il trattamento è con dosaggi che sono anche 10 volte superiori alla DL.
Altre strategie per antagonizzare un ligando sono la formazione di un legame irreversibile sul recettore, l’azione su un sito diverso da quello dell’agonista o ad un livello successivo.
Caso di antagonista competitivo irreversibile
(esemplificazione: relazione diretta tra occupazione recettoriale ed effetto)
B
+ la nuova reazione influenza Rt
A + R ↔ AR con 0 ≤ q ≤ 1
↓
BR
AR = Rt AR= Rt • q
1 + Ka/A 1 + Ka/A
L’antagonista competitivo irreversibile influenza l’efficacia dell’agonista, oltre che la sua potenza. Solitamente però si ha un numero di recettori esuberante rispetto al numero necessario per raggiungere l’effetto massimale. Se l’antagonista non reversibile occupa un numero di recettori minore rispetto a quelli necessari per l’effetto massimale si ha sempre l’effetto massimo. In questo caso inizialmente l’antagonista sposta la curva come un antagonista reversibile. Secondo il modello di Schild uno spostamento di questo tipo non obbedisce a un equilibrio simultaneo.
Eff = ƒ(AR)
Data una coppia di concentrazioni equiattive si ha un certo numero di recettori occupati che risulta uguale per l’ottenimento di un effetto uguale.
Eff = ƒ Rt Eff = ƒ Rt • q
1 + Ka/A 1 + Ka/A’
ƒ Rt = ƒ Rt • q ƒ è inconoscibile
1 + Ka/A 1 + Ka/A’ ma uguale nei due casi
Rt = Rt1 • q
1 + Ka/A 1 + Ka/A’
1 = 1
1 + Ka/A 1/q + Ka/A’q
1 + Ka = 1 + Ka
A q A’q
1 1 + 1 1 = 1 + 1
q Ka A’ q Ka A
1= 1 1 + 1 1 – 1
A A’ q Ka q
L’equazione che si ottiene è quella di una retta con pendenza S = 1/q e intercetta i = 1/Ka • [(1/q)-1]
da ciò si ricava che Ka = (S-1)/i
La risposta al farmaco agonista
Esistono risposte che per loro natura sono quantali oppure risposte graduali che si possono trattare, per comodità, come quantali.
In questo caso la risposta farmacologica ha un andamento gaussiano.e la gaussiana viene analizzata in modo cumulativo si trasforma in una sigmoide che varia in base all’incidenza di effetto su una data popolazione. La dose per cui risponde il 50% della popolazione esaminata è detta ED50. Si può costruire anche la curva della DL50, il rapporto tra le due consente di stimare l’indice terapeutico IT=DL50/DE50 e il margine di sicurezza (DL1/DE99).
Un recettore è una macromolecola che accetta il legame (selettivo), a livello cellulare, da parte di un ligando. Il recettore attivato deve far scaturire un effetto biologico o farmacologico.
La classificazione dei recettori può essere fatta in base al luogo della cellula in cui si trovano: quindi si avranno recettori intracellulari e recettori di membrana; i recettori intracellulari sono regolatori della trascrizione genica e possiedono tutti un’analogia strutturale e funzionale; i recettori di membrana sono invece estremamente dissimili tra loro.
Recettori di membrana
I recettori di membrana possono essere divisi in:
- metabotropici, la loro attivazione è seguita da meccanismi cellulari caratterizzati da reazioni chimiche;
- ionotropici, associati a canali ionici, la loro attivazione consente il transito di specie ioniche.
I recettori intracellulari sono proteine citosoliche o nucleari che presentano sequenze di 400-1000 aminoacidi; caratterizzati da tre parti distinte: la porzione carbossiterminale, con un sito di legame per il ligando endogeno; la porzione intermedia, che definisce la specificità di legame con una determinata sequenza di DNA e zona in cui è legata una proteina regolatoria (HSP), con funzione inibitoria, in condizioni di riposo; la porzione amminoterminale, che permette l’inizio della regolazione dell’espressione genica.
Come si attiva il recettore intracellulare?
Il recettore si attiva tramite il legame con il mediatore che, legandosi, produce una modifica allosterica che fa sì che si abbia perdita di affinità per la HSP; il complesso recettore-mediatore dimerizza e va a produrre l’effetto. I siti del DNA in cui il complesso produce l’effetto sono composti da sequenze palindrome (HRE); all’interno del nucleo i recettori attivati si legano alle HRE e scatenano una stimolazione di tipo regolatorio.
Le modifiche indotte da un mediatore su questo tipo di recettore sono a lenta azione ma durano a lungo.
Su recettori di questo tipo si interviene con farmaci che mimano l’azione dei mediatori, quindi agonisti; se la via è eccessivamente attivata si può ricorrere all’uso di un antagonista, molecola che occupa il sito di legame del mediatore ma impedisce l’attività del ligando. Gli antagonisti si dividono in due categorie: antagonisti di tipo 1, si legano al sito recettoriale senza che conseguano altre azioni; antagonisti di tipo 2, si legano al recettore e lo attivano ma le modifiche non rendono attiva la parte amminoterminale del recettore.
Recettori metabotropici e proteina G
I recettori metabotropici di membrana accoppiati a proteinaG hanno tutti un’analogia strutturale: sono costituiti da un unico filamento proteico che attraversa la membrana sette volte; il loop tra il V° e il VI° dominio è accoppiato alla proteinaG; il sito di legame con il mediatore è sul lato esterno della membrana. La proteinaG è un complesso proteico formato da tre subunità (α, β e γ) di cui la α è la più grande e presenta un sito di legame per il GDP/GTP. In condizioni basali il GDP è legato alla subunità α, quando arriva il mediatore si ha un cambiamento di conformazione del recettore che induce una modifica allosterica sulla proteinaG, ciò induce il distacco della subunità α e il GDP viene rimpiazzato dal GTP; la subunità attivata ha affinità per un effettore a valle.
- Gs – regolazione positiva, l’effettore è l’adenilato-ciclasi.
- Gi – regolazione inibitoria, l’effettore è l’adenilato-ciclasi.
- Gq – innesca meccanismi inositidici, l’effettore è la fosfolipasiC.
- Go – accoppiata a proteine che interagiscono con canali ionici d’entrata.
Il sito di legame per il GTP non è passivo ma ha attività catalitica GTPasica, quindi è in grado di mediare reazioni chimiche. L’estensione del segnale per il cAMP è mediata dalla fosfodiesterasi (PDE), che pone termine all’effetto iniziato con la stimolazione del recettore primario.
Il cAMP può attivare le PKA (proteinchinasi-A) che andranno a fosforilare tutta una serie di target.
Un altro effettore delle proteineG (Gq) è la fosfolipasiC (PLCβ); questa molecola innesca un meccanismo più complesso di quello prodotto dall’adenilato-ciclasi: una volta attivata dalla proteinaG va ad intervenire su particolari fosfolipidi di membrana, fosfatidil inositolo difosfato (PIP2) provocandone l’idrolisi.
Le PLC idrolizzano il legame tra l’inositolo difosfato e il glicerolo, formando inositolo trifosfato (IP3) e diacilglicerolo (DAG). Il DAG rimane sulla membrana, non come costituente inerte, ma assume il significato di secondo messaggero interagendo con una proteinchinasi (PKC) che attiva una cascata fosforilativa.
La risposta rapida è però data dall’IP3 che va ad agire su un canale situato sul reticolo endoplasmatico consentendo la fuoriuscita di ioni Ca++, che vi erano immagazzinati.
Il calcio liberato dal reticolo endoplasmatico promuove l’attivazione di vie di membrana per l’ingresso di altro calcio nella cellula.
Effetti possibili
Ci sono almeno due effetti possibili:
- se la cellula è eccitabile l’alterazione della concentrazione di calcio porta ad un’alterazione del potenziale di membrana che determina l’apertura di canali voltaggio-dipendenti;
- se la cellula non è eccitabile, al posto dei canali voltaggio-dipendenti, esistono canali CRAC (calcium release activated channels). Anche il calcio può attivare le proteinchinasi.
I recettori a tirosinchinasi sono una classe meno importante, rispetto alle altre, dal punto di vista quantitativo ma ugualmente importanti per la loro funzione.
Questi recettori sono caratterizzati da una struttura comune: un unico filamento trans-membrana formato da una o due proteine, di cui una subunità completamente esterna. All’esterno si ha la presenza di un sito di riconoscimento per il mediatore endogeno e all’interno un sito catalitico.
Quando arriva il mediatore si ha il legame con il recettore e la dimerizzazione, unione di un’altro recettore al primo complesso, che innesca l’attivazione della proteina presente nel dominio catalitico.
Recettori canale
I recettori canale sono tra le possibili tipologie di canali ionici.
La membrana per sua natura e funzione è una barriera che presenta una quasi-totale impermeabilità alle specie idrofile ed agli elettroliti. Però le specie ioniche devono poter transitare; la permeazione passiva è prossima allo zero, per questo ci sono proteine trans-membrana con questo compito.
Le pompe operano un flusso a discapito di energia (trasporto attivo contro gradiente), sono proteine ATPasiche (Na+/K+ ATPasi) che usano il 30-70% dell’ATP prodotta; ciò fa sì che la concentrazione di potassio sia circa 140mM all’interno della cellula e 5mM all’esterno, mentre quella del sodio 10mM all’interno e 150mM all’esterno.
Antiporter o co-trasportatori
Un’altra via di transito trans-membrana è ad opera di trasportatori antiporter o co-trasportatori:
- un antiporter è uno scambiatore, come l’antiporto Na+/Ca++, che sfrutta il gradiente del sodio in ingresso per far uscire il calcio;
- nel co-trasporto due ioni entrano insieme, uno secondo gradiente e l’altro perché ha un sito di legame sullo stesso canale.
Nei canali ionici gli ioni passano secondo gradiente.
Ca++ ~ μM → ~ mM
Cl– 5-15mM → 100-150mM
Se la permeabilità è poca ma presente si può
creare una differenza di potenziale sulla membrana.
Equazione di Nerst:
E = RT · ln [X]ext
zf [X]int
barriera impermeabile
per una membrana biologica si ha:
E = +/- 60 · log10 [X]ext
[X]intImponendo un potenziale alla membrana l’equilibrio tra le concentrazioni dello ione si ristabilisce in base all’equazione di Nerst.
Nelle nostre cellule non è presente però un solo ione che determina la differenza di potenziale della membrana, ma ogni specie ionica determina un suo potenziale di equilibrio.
Il reale potenziale di membrana è circa -60/-80mV (all’interno) e deriva dalla somma ponderata di tutti i potenziali di equilibrio dei vari ioni; ogni ione presenta una conduttanza specifica, dovuta alla permeabilità dello ione stesso. Il potenziale di membrana è quindi influenzato principalmente dalla permeabilità. Se si aumenta la conduttanza a una specie ionica il potenziale di membrana tende verso quello della specie ionica in questione.
Canali ionici
Esistono meccanismi biologici in grado di consentire un aumento specifico della permeabilità di membrana, sono i canali ionici.
I canali ionici si possono distinguere in:
- Recettori-canale (ionotropici) regolati da ligandi specifici extracellulari; sono proteine di membrana che si aprono o chiudono in relazione al legame con una proteina.
- Voltaggio-regolati; risentono degli spostamenti del potenziale di membrana.
Una depolarizzazione della membrana corrisponde a un’eccitazione cellulare, la polarizzazione favorisce la stabilizzazione: quindi l’apertura di un canale per il Na+ determina un’eccitazione mentre uno per il K+ determina una stabilizzazione; il Cl– si muove in base al potenziale di membrana, se positivo entra, se negativo esce, in entrambi i casi si stabilizza la cellula.
Recettori inotropici
I recettori ionotropici presentano tutti un’analogia nella composizione strutturale: sono eteropentameri costituiti da cinque proteine trans-membrana che si raggruppano a formare un fascio. Ogni proteina è costituita da quattro domini a α-elica trans membrana (M1, M2, M3, M4), le due estremità sono extracellulari. Il segmento M2 è ricco di aminoacidi idrofilici, si affaccia verso il poro e permette il passaggio degli ioni.
Recettore nicotinico
Nel recettore nicotinico la porzione extracellulare che esprime il sito recettoriale è sulla proteina α. Il legame sulla α fa sì che il pentamero cambi conformazione, si apra e permetta il passaggio di alcuni ioni.
Un’altra categoria di recettori-canale presenta ligandi intracellulari quali metaboliti o specie ioniche, per esempio si hanno canali di K+ inibiti dall’ATP, attivati da Ca++ o Cl–.
I canali ionici voltaggio-dipendenti e con ligandi intracellulari presentano una selettività molto superiore rispetto ai canali ionotropici. La regolazione dei recettori ionotropici è data solamente dalla dimensione del poro.
Canali voltaggio-operati
I canali voltaggio-operati sono i più differenziati e ampiamente espressi; esistono per tutte le tipologie di elettroliti. A dispetto della specificità presentano tutti una struttura analoga: sono costituiti da quattro proteine (tetramero) che possono essere uguali o diverse, ma sono tutte α; spesso presenta subunità accessorie che vanno a modulare l’attività del canale. Ogni proteina α è formata da sei domini, con estremità esterne, trans membrana (S1-S6). Il loop S5-S6 è la porzione della catena che si affaccia verso il poro; dominio S4 presenta il meccanismo che rende il canale sensibile al voltaggio. Nella sequenza amminoacidica del dominio S4 è presente un’arginina (+) ogni tre aminoacidi; per attrazione elettrostatica le arginine sono attratte da cariche relativamente negative poste verso l’esterno, ma il potenziale negativo
interno attrae le cariche internamente, stabilizzandole. Se il potenziale viene reso positivo la forza di attrazione verso
l’interno è inferiore a quella verso l’esterno e il canale si apre. Il potenziale di attivazione dei canali si aggira sui -55mV; l’effetto dipende dal tipo di canale, si può avere un’ulteriore depolarizzazione o un feedback. Il loro andamento dipende dalle cinetiche di attivazione dei canali.
I canali che sono anche recettori possono essere modulati farmacologicamente come tutti gli altri recettori, usando sostanze agoniste (mimetiche), antagoniste e anche sostanze agoniste ma con un effetto inattivante.
I canali voltaggio-operati non hanno la possibilità di essere modulati da agenti chimici, la loro attività è condizionata dal voltaggio della membrana; la soglia intrinseca di attivazione può essere modificata da fosforilazioni o proteine accessorie. Esistono sostanze che agiscono su questo tipo di canali ionici inducendo alterazioni conformazionali che modificano la soglia di sensibilità dei canali; sostanze che abbassano la soglia di attività sono definite attivatori, da sole non sono in grado di consentire l’apertura del canale; i modulatori negativi aumentano la soglia di attivazione mentre i bloccanti occupano fisicamente il poro impedendo il transito degli ioni.
L’organismo ha una vastissima serie di sostanze che agiscono come agonisti verso i loro recettori con una reciproca selettività e presenta una continua regolazione (prossimale) di questi recettori. Si possono osservare principalmente due tipi di regolazione: positiva, rappresenta eccezioni, e negativa, reprime l’attività recettoriale.
Cos’è la tolleranza nei sistemi recettori?
La capacità dei sistemi recettoriali di ridurre la stimolazione ha implicazioni evidenti. La principale forma in cui si manifesta un meccanismo di questo tipo è nota come tolleranza. La tolleranza, in generale, è la perdita di efficacia di una data dose di farmaco, che produce, man mano che il farmaco viene utilizzato, una diminuita risposta incrementabile con l’aumento del dosaggio. La tolleranza riguarda però tutti gli effetti del farmaco.
Quando i meccanismi di tolleranza attivati dal farmaco riguardano l’attività del farmaco sul recettore si parla di tolleranza farmacodinamica; la tolleranza farmacocinetica è legata al metabolismo del farmaco.
La tolleranza farmacodinamica può instaurarsi nel tempo oppure in tempi molto brevi, legata a meccanismi molecolari (tachifilassi).
Effetto rebound
Un altro problema derivante dalla tolleranza è il cosiddetto effetto rebound, o effetto rimbalzo; l’organismo si adatta alla presenza di un farmaco che se sospeso bruscamente porta a un brusco effetto opposto, talvolta più grave della malattia stessa. Per questo la sospensione deve avvenire in maniera graduale. La tolleranza induce dipendenza. L’organismo risponde alla mancanza del farmaco provocando sintomi gravi e talvolta potenzialmente mortali.
La desensitizzazione, ossia la modulazione negativa, cambia a seconda dei meccanismi e del tipo di recettori coinvolti. Quindi la desensitizzazione dei recettori ionotropici è molto diversa rispetto a quella dei recettori accoppiati a proteinaG.
Per quanto riguarda il recettore ionotropico il meccanismo di desensitizzazione risiede in una proprietà del canale stesso, sia esso recettoriale o voltaggio-operato. Il canale può esistere in tre conformazioni diverse che sono: a riposo, attivo e inattivo.
Nel canale voltaggio-operato del calcio (tipo L) agiscono i calcio-antagonisti, bloccano il poro del calcio. Si hanno tre diverse categorie di questi farmaci: diarilalchilammine (verapamil), benzotiadiazine (diltiazem), diidropiridine (nicardipina, ecc). Hanno tutti una simile affinità per il sito di legame; le prime due hanno una modalità di legame con il canale frequenza-dipendente, sebbene l’affinità sia la stessa si legano tanto meglio quanto più è frequente il ciclo, mentre l’altra è stato-dipendente, si lega tanto meglio quanto più il canale esiste nella forma attivata.
Recettore cationico
Il recettore cationico nicotinico presenta un sito di legame per l’acetilcolina; in questa coesistenza di tre stati conformazionali l’acetilcolina sposta l’equilibrio verso la forma attivata, il canale transita verso lo stato di inattivazione e poi in quello di riposo, con la dissociazione del ligando dal suo sito recettoriale. Alcuni agonisti non si dissociano dal recettore, quindi non si ha il ritorno allo stato di riposo e il recettore non è più in grado di attivarsi.
Per i recettori accoppiati a proteina G, per esempio i beta-adrenergici, si ha una vasta gamma di meccanismi di desensitizzazione. Le subunità β e γ della proteinaG, dissociate dalla α, vanno ad interagire con una proteinchinasi chiamata βARK (chinasi del recettore beta-adrenergico), fosforilandola. Questa chinasi si attiva e va a fosforilare il loop tra i domini V e VI, zona di contatto tra la proteinaG e il recettore. Ciò consente al loop di acquisire affinità e legare la beta-arrestina. La beta-arrestina provoca una modifica conformazionale del sito, che rende meno affine il recettore per l’agonista; anche la proteinchinasi cAMP-dipendente attua una fosforilazione sul recettore, innescando un meccanismo di desensitizzazione che origina dalla stessa trasduzione del segnale.
La desensitizzazione omologa è specifica per un tipo di recettore; mentre un aumento di cAMP, dovuto anche a fattori indipendenti, porta comunque a una desensitizzazione, in questo caso eterologa.
Come ulteriore meccanismo di desensitizzazione si può avere, attraverso meccanismi di fosforilazione PKA-mediati, l’internalizzazione della porzione di membrana che presenta il recettore, fino ad arrivare al distacco della vescicola contenente il recettore, con formazione di un endosoma. (down regulation recettoriale). Successivamente si può avere la defosforilazione, con ritorno del recettore sulla superficie, o la proteolisi e la degradazione.
Si ha anche una down regulation tardiva, di origine genica, anche a carico delle subunità della proteinaG o le ciclasi.
Per quanto riguarda i recettori tirosinchinasici la desensitizzazione è probabilmente dovuta a meccanismi fosforilativi che limitano la capacità del recettore; invece la desensitizzazione dei recettori intracellulari risiede in un meccanismo genico, con un impoverimento del numero di recettori.
