esistono diverse tipologie di alterazioni sessuali, a seconda se di origine genetica, familiare o altro, vediamo insieme quali sono:
• Disgenesia dei tubuli seminiferi: Sindrome di Klinefelter
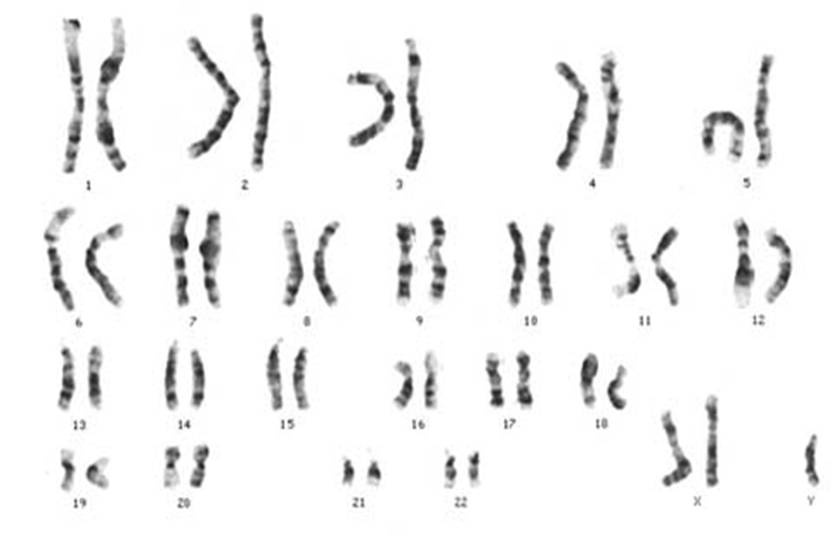
Essa rappresenta una delle forme più frequenti di ipogonadismo primario e di sterilità maschile.
Genotipo XXY (presenza di un cromosoma X in più nel corredo genetico): dovuto ad una non-disgiunzione meiotica materna, che dà luogo, quindi, ad un ovulo con due cromosomi X (genitori anziani ).
esistono diverse tipologie di alterazioni sessuali, a seconda se di origine genetica, familiare o altro, vediamo insieme quali sono:
• Disgenesia dei tubuli seminiferi: Sindrome di Klinefelter
Essa rappresenta una delle forme più frequenti di ipogonadismo primario e di sterilità maschile.
Genotipo XXY (presenza di un cromosoma X in più nel corredo genetico): dovuto ad una non-disgiunzione meiotica materna, che dà luogo, quindi, ad un ovulo con due cromosomi X (genitori anziani ).
Segni clinici: nessun sintomo prima della pubertà. Alla pubertà si osserva: fibrosi ed ialinizzazione tubuli seminiferi; testicoli piccoli e duri; cellule del Leydig disposte in ammassi ed iperplasiche (volume normale → riduzione volume tubuli seminiferi); ↓ livelli di testosterone, ↑ gonadotropine; ginecomastia; azoospermia (chiusura tubuli seminiferi); scarsa libido; ridotta potenza; proporzioni scheletriche anormali (estremità inferiori maggiori di quelle superiori; apertura delle braccia superiore all’altezza); deficit intellettivo e comportamento asociale.
Maggiore frequenza di alcune condizioni patologiche: malattie polmonari croniche, varici venose, intolleranza al glucosio, ipotiroidismo primario, rischio maggiore di cancro alla mammella.
Varianti della Sindrome di Klinefelter: aumentato numero di cromosomi X e Y e mosaicismi 46,XY/47,XXY. Maggiore frequenza di anomalie dello sviluppo (sinostosi radio-ulnare) e ritardo mentale.
Maschio 46XX: fenotipo maschile, aspetti istologici simili a quelli con cariotipo 47,XXY, proporzioni corporee normali, statura media più bassa rispetto a quelli con cariotipo XXY o ai maschi normali.
• Sindrome della disgenesia gonadica: Sindrome di Turner
Genotipo 45,X o XO, fenotipo femminile
Segni clinici:
– anomalie somatiche (linfedema delle estremità, micrognatia, pieghe epicantiche, orecchie sporgenti ed attaccatura bassa, bocca a pesce e ptosi, torace a forma di scudo, collo corto)
– infantilismo sessuale all’epoca della pubertà secondario a disgenesia gonadica (gonadi atrofiche, stroma fibroso), ↑ gonadotropine (assenza inibizione a feedback dell’asse ipotalamo-ipofisi-gonade)
– statura bassa
– coartazione dell’aorta (10%), ipertensione
– malformazioni renali
– nevi pigmentati
– dorso delle mani paffuto, deformità del polso, scoliosi
– frequenti otiti dell’orecchio medio
Maggiore frequenza di: obesità, osteoporosi, diabete mellito, tiroidite di Hashimoto, artrite reumatoide, enteropatie infiammatorie, anoressia nervosa.
Trattamento a base di estrogeni, per indurre la manifestazione dei caratteri sessuali secondari ed il menarca.
• Disgenesia gonadica 46,XX e 46,XY: gonadi atrofiche bilaterali, fenotipo femminile, assenza delle caratteristiche della sindrome di Turner, infantilismo sessuale, statura normale o alta e proporzioni eunucoidi.
• Ermafroditismo vero
Presenza in una o entrambe le gonadi, di tessuto ovarico e testicolare
Maggiore frequenza di ovotestis, ovaio ed, in misura minore testicolo.
Ovotestis: ovaio e componente ovarica → normale funzionamento; testicolo → disgenetico
Cariotipo 46,XX (60%), 46,XY (20%), mosaicismo o chimerismo 46,XX/46,XY
La maggioranza dei veri ermafroditi 46,XX risulta SRY-negativa.
