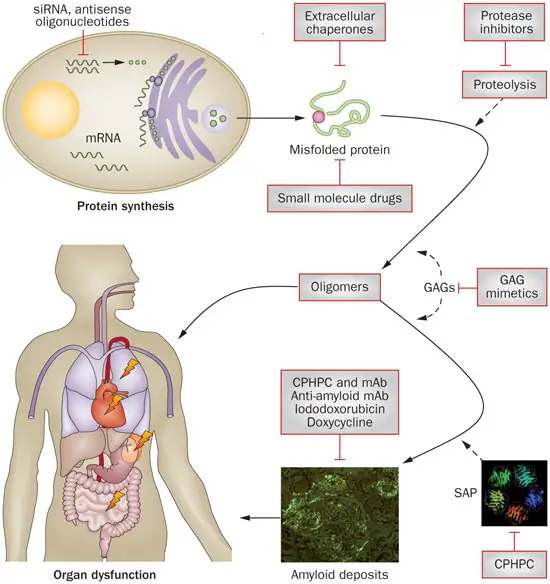
Con l’allungamento dell’aspettativa di vita le patologie degenerative ed in particolare la forma di amiloidosi che si ha a carico del SNC (morbo di Alzheimer) sono diventate una grave malattia sociale.
L’amiloidosi può essere la conseguenza di un’infiammazione cronica ed è dovuta alla deposizione di materiale amorfo di natura proteica in sede extracellulare. L’organo prevalentemente colpito dipende dal tipo di amiloidosi.
Con l’allungamento dell’aspettativa di vita le patologie degenerative ed in particolare la forma di amiloidosi che si ha a carico del SNC (morbo di Alzheimer) sono diventate una grave malattia sociale.
L’amiloidosi può essere la conseguenza di un’infiammazione cronica ed è dovuta alla deposizione di materiale amorfo di natura proteica in sede extracellulare. L’organo prevalentemente colpito dipende dal tipo di amiloidosi.
Il termine amiloidosi deriva da amido perché i patologi della II metà dell’800 notarono che c’erano delle condizioni morbose in cui si notava una sostanza in sede extracellulare con proprietà simili a quelle dell’amido (metacramasia).
Trattando il tessuto con un particolare tipo di colorante, il rosso Congo, si colora questa sostanza in maniera selettiva. Il colorante può essere iniettato in un paziente e si può poi misurarne la clearance: se ci sono depositi amiloidi una parte di questo indicatore viene sequestrato e la clearance diminuisce.
Su base clinica le amiloidosi possono essere distinte in primarie o secondarie a seconda che la malattia sia senza cause apparenti o se, viceversa, accompagni altre patologie.
Questi depositi possono provocare danni funzionali.
Per quando riguarda la natura dell’amiloide, i depositi sono da un punto di vista della loro componente molecolare così formati:
· Circa il 90% è costituito da una componente proteica specifica che tende a depositarsi in forma di fibrille. Questa componente varia a seconda del tipo di amiloidosi ed è la responsabile delle proprietà tintoriali;
· Circa il 10% è costituito da una componente costante P (serum amyloid P component, SAP): è una proteina sierica appartenente al gruppo delle pentrassine. Si lega alle fibrille in tutte le forme di amiloidosi, ad eccezione dell’Alzheimer;
· Una piccola quantità è costituita da glicosaminoglicani.
Per molti anni si è classificato le amiloidosi come malattie primarie. Poi ci si è resi conto che il fenomeno è complesso e si è cominciato a classificare le amiloidosi in base al tipo di proteina specifica che si deposita.
Le principali amiloidosi sono:
1. AL: è legata a proliferazione monoclonale dei linfociti B. Colpisce in stragrande maggioranza pazienti affetti da mieloma multiplo. E’ causata da un’iperproduzione di catene leggere, soprattutto di tipo λ, rispetto a quelle pesanti. Ciò da una parte provoca proteinuria e dall’altra porta alla creazione di depositi amiloidi;
2. AA: sono conseguenza di malattie croniche. Si depositano proteine dette SAA, appartenenti alle proteine di fase acuta;
3. compare in pazienti dializzati ed è dovuta al fatto che un componente dell’HMC, la β2 microglobulina, tende a depositarsi;
4. di tipo genetico legata a mutazioni puntiformi della prealbumina, porta ad una certa facilità di aggregazione e di deposito nei tessuti;
5. forme endocrine: particolarmente nota è l’amiloidosi che si accompagna al diabete di tipo II dove si ritrovano depositi amiloidi all’interno delle isole pancreatiche. Questi depositi dipendono dalla secrezione da parte del pancreas di un piccolo peptide, l’amilina, co-secreto con l’insulina. Si è scoperto che l’amilina è una proteina che in vitro ha effetti anti-insulinici: si è perciò pensato che il diabete di tipo II potesse essere dovuto ad un’iperproduzione di amilina. Oggi l’ipotesi è tramontata e il ruolo biologico di questo peptide resta ancora da definire;
6. MALATTIA DI ALZHEIMER: si tratta di una malattia di lunga durata che compare all’inizio con dei sintomi molto vari (disturbi dell’umore, perdita di memoria, disorientamento temporale e spaziale) e perciò per la diagnosi sono necessari almeno 1/2 anni dall’insorgenza.
Con l’aggravarsi della malattia si notano segni caratteristici:
– amnesia anterograda;
– disturbi del comportamento;
– col progredire della malattia si arriva alla demenza completa.
Tra un cervello di un paziente normale e quello di uno affetto le differenze sono notevoli: vi è una forte atrofia corticale con ingrandimento dei ventricoli conseguentemente alla perdita di parenchima (neuroni colinergici).
Alla PET si vede chiaramente che un cervello di un paziente Alzheimer è più piccolo e l’utilizzazione del glucosio è fortemente ridotta.
In diverse zone cerebrali vi è la deposizione di materiale amorfo con una particolare struttura: un core centrale che sembra essere l’amiloide circondato da filamenti a raggio risultato della degenerazione degli assoni e dei dendriti dei neuroni circostanti.
I depositi sono dovuti ad un piccolo peptide di 42 AA, detto β42, il quale è contenuto normalmente su una proteina di superficie dei neuroni (APP: amyloid precursor protein). La proteina è tagliata da un gruppo di proteasi e solo questo prodotto è responsabile della malattia.
Può succedere che la proteina APP normale vada incontro a proteolisi selettiva da parte delle α,β, e γ secretasi. A seconda della via di taglio può o meno instaurarsi la malattia: se la proteina è tagliata dalle α secretasi non si verifica malattia, ma se essa è tagliata prima dalle β e poi dalle γ si verifica malattia.
Circa il 5% dei pazienti affetti da Alzheimer hanno una predisposizione familiare: evidentemente ci sono delle alterazioni genetiche dell’APP oppure nei geni modulatori della funzione secretasica.
Le forme familiari sono caratterizzate da una precoce età di insorgenza (prima dei 65 anni) e da un’ereditarietà autosomica dominante.
Nel resto dei casi si parla di Alzheimer sporadico e non si è in grado di capire la patogenesi.
