L’infiammazione cronica è caratterizzata dalla presenza di una guaina fibrosa atta a contenere il sito infiammato.
Se però la produzione di tessuto fibroso da parte dei fibroblasti non è controllata, può evolvere in fibrosi.
Le cause della fibrosi possono essere:
citochine prodotte da macrofagi, linfociti-T, plasmacellule, piastrine, piastrine, eosinofili e mastcellule.
Serotonina provoca fibrosi a livello del miocardio.
Silicio, asbesto e tutto il materiale inerte.
Ischemia.
L’infiammazione cronica è caratterizzata dalla presenza di una guaina fibrosa atta a contenere il sito infiammato.
Se però la produzione di tessuto fibroso da parte dei fibroblasti non è controllata, può evolvere in fibrosi.
Le cause della fibrosi possono essere:
citochine prodotte da macrofagi, linfociti-T, plasmacellule, piastrine, piastrine, eosinofili e mastcellule.
Serotonina provoca fibrosi a livello del miocardio.
Silicio, asbesto e tutto il materiale inerte.
Ischemia.

E’ la più comune malattia autosomica recessiva nella popolazione caucasica: 1 persona su 25 è eterozigote e l’incidenza della malattia è di 1/2500. L’eterozigote è una persona del tutto normale.
La malattia è monogenica: nel 1980 è stato dimostrato che il tessuto epiteliale negli organi colpiti dalla malattia è impermeabile al cloro e nel 1989 è stato isolato il gene per la fibrosi cistica, chiamato CFTR (regolatore di conduttanza transmembrana della FC) mentre nel 1991 si è scoperto che la proteina prodotta da CFTR è un canale che trasporta lo ione cloro.
CFTR: E’ una proteina transmembrana, localizzata nella porzione apicale delle cellule epiteliali, appartenente alla superfamiglia dei trasportatori ABC (ATP binding casset).
La proteina è costituita da due domini transmembrana che definiscono la selettività del canale e da due domini NBD che idrolizzano ATP. Infine è presente un dominio R, regolatorio, che quando viene fosforilato da una PkA apre il canale.
CFTR subisce una maturazione post-traduzionale nel Golgi che consiste in una glicosilazione e nel “controllo qualità”. Esistono numerose diverse mutazioni a carico del gene CFTR. Sebbene ne siano state individuate centinaia esse possono essere raggruppate in 4 classi:
1. Alterata produzione della proteina a causa di mutazioni geniche di svariati tipi;
2. Alterazione del processo di maturazione post-traduzionale (es. per anomala glicosilazione);
3. Alterazione dei domini regolatori: in genere sono mutazioni a carico dei siti di legame per l’ATP o a livello dei siti regolatori;
4. Mutazioni nei domini transmembrana che determinano un’alterata conduttanza del canale.
Nei primi due casi la proteina viene degradata e di fatto viene a mancare la proteina canale a livello apicale.
La mutazione di gran lunga più frequente (66%) è una delezione di tre basi (detta F508, dove F sta per fenilalanina). La delezione porta, come risultato finale, alla mancanza di una fenilalanina in posizione 508. In alcune zone questa mutazione rappresenta la causa del 90% dei casi di fibrosi cistica, mentre in Italia la percentuale scende al 52%.
La mutazione causa una glicosilazione anomala e comporta un anomalo ripiegamento. Il sistema “controllo qualità” riconosce l’anomalia e induce la degradazione della proteina nel proteosoma. Il risultato finale è la mancanza del canale del cloro.
Patologia
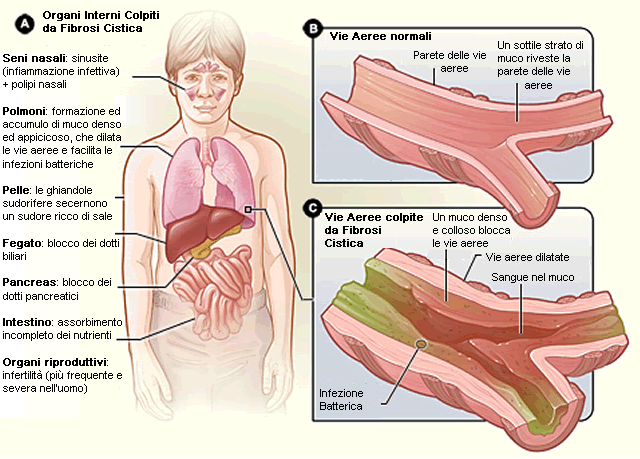
La secrezione del cloro è fondamentale per garantire l’adeguata idratazione delle mucose. Gli organi più colpiti sono i polmoni, il pancreas, l’apparato gastroenterico, l’apparato genito-urinario e le ghiandole sudoripare. La sintomatologia è molto variabile: può essere piuttosto lieve oppure grave e può insorgere alla nascita (5-10% dei casi) o anni dopo. Questa variabilità è presente anche all’interno di uno stesso genotipo ma non si sa bene perché.
Se manca FCTR vi è assorbimento di sodio a livello delle mucose ma non secrezione di cloro con una conseguente disidratazione per effetto osmotico.
Nelle ghiandole sudoripare vi è riassorbimento simporto Na/Cl. Se manca il trasportatore per il cloro il sudore è molto più ricco di ioni. Se infatti si dosano gli elettroliti nel sudore si osserva che la concentrazione è di 40-50 mEq/l nei soggetti normali mentre essa sale a 70 mEq/l nei soggetti affetti da fibrosi cistica.
Segni clinici
· Vie aeree: presenza di muco denso e viscoso che provoca ostruzione e inibisce il movimento cigliare dell’epitelio polmonare. Ciò provoca difficoltà respiratorie e predisposizione a infezioni che sono la causa della gran parte delle morti da FC (streptococcus aureus e pseudomonas aeruginosa, quest’ultimo trova un ambiente di crescita ideale nel muco viscoso presente in questi pazienti). Le infezioni portano a distruzione del parenchima e conseguente fibrosi;
· Fegato: il blocco dei dotti biliari compromette la digestione e la funzionalità è ridotta. Questo succede però solo nel 5% dei casi;
· Pancreas: l’occlusione dei dotti, nell’85% dei pazienti, impedisce al pancreas di liberare enzimi nel tubo digerente e causa atrofia e fibrosi del pancreas esocrino;
· Intestino tenue: l’occlusione del canale intestinale da parte di feci compatte richiede l’intervento chirurgico in circa il 10% dei neonati (ilo da meconio);
· Apparato riproduttivo: l’assenza dei dotti deferenti (causa sconosciuta) rende sterile il 95% dei maschi. Anche le donne possono essere sterili in seguito alla formazione di un tappo mucoso che impedisce l’ingresso del liquido seminale nell’utero;
· Cute: alte concentrazioni di elettroliti nel sudore.
Terapia
La terapia genica, ad oggi, per questa malattia non ha avuto successo perché non si sono trovati vettori adeguati. Sono in corso di studio dei farmaci che “scortino” le proteine canale che, anche se glicosilate in maniera anomala, sarebbero funzionali. Un’altra strategia terapeutica è quella di potenziare altre classi di canali del cloro.
I pazienti devono essere sempre tenuti sotto controllo perché vanno spesso incontro ad infezioni e l’uso di antibiotici ed anti-infiammatori è quasi costitutivo.
