Nelle cellule neoplastiche le mutazioni a carico dei geni oncosoppressori devono sempre colpire entrambi gli alleli. La mutazione di uno solo dei due alleli difatti normalmente non dà origine a tumori, anche se non sempre passa inosservata. Ciò è dovuto al fatto che la mutazione di un oncosoppressore provoca perdita di funzione la quale può essere compensata dall’espressione dell’altro allele. Si parla perciò di mutazione con effetto recessivo.
Nelle cellule neoplastiche le mutazioni a carico dei geni oncosoppressori devono sempre colpire entrambi gli alleli. La mutazione di uno solo dei due alleli difatti normalmente non dà origine a tumori, anche se non sempre passa inosservata. Ciò è dovuto al fatto che la mutazione di un oncosoppressore provoca perdita di funzione la quale può essere compensata dall’espressione dell’altro allele. Si parla perciò di mutazione con effetto recessivo. La mutazione di un solo oncogene invece è sufficiente per rappresentare un guadagno di funzione: si parla allora di mutazione con effetto dominante.
Le mutazioni possono essere qualitative (puntiformi) o quantitative (perdita di materiale genetico o inattivazione dei prodotti proteici). Un esempio di mutazione quantitativa è la LOH (loss of eterozigosity)
A differenza di quanto avviene per gli oncogeni la mutazione degli oncosoppressore può colpire le cellule germinali ed essere trasmessa alla prole. I figli saranno maggiormente esposti al rischio di sviluppare tumore essendo sufficiente una singola mutazione (sull’allele sano).
Alcuni oncogeni possono essere trasmessi per via ereditaria:
met: provoca carcinoma papillare del rene;
ret: sindrome da neoplasia endocrina multipla (MEN tipo 2);
Carcinoma midollare familiare della tiroide;
Morbo di Hirschspring (o mega-colon congenito).
Alcuni oncosoppressori, contravvenendo alla regola, hanno invece effetto dominante. Un esempio è P53 che deve formare degli omotetrameri per funzionare. Se solo uno dei 4 monomeri è alterato l’intero tetramero non funziona più.
Ci sono tre osservazioni che giustificano quanto detto in precedenza:
· Osservazione sperimentale: se si crea un ibrido dalla fusione di una cellula tumorale (due alleli malati) con una cellula sana (due alleli sani) la cellula risultante non avrà fenotipo tumorale perché i prodotti dei geni sani sono in grado di compensare. Se però uno dei cromosomi sani viene eliminato allora la cellula assume fenotipo neoplastico. Questo fenomeno è detto “aploinsufficienza”: il prodotto di un singolo allele sano a volte non è sufficiente per ripristinare la normale funzionalità cellulare;
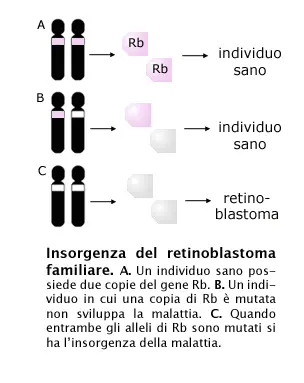
· Osservazione clinica: nel retinoblastoma familiare c’è un gene, chiamato RB, che subisce la mutazione in uno dei due alleli già in fase pre-zigotica. Ne consegue che tutte le cellule somatiche hanno un allele RB mutato ed uno normale. Se per cause successive si verificano altre mutazioni anche l’altro gene RB può essere alterato. La cellula assume così fenotipo neoplastico.
Nel retinoblastoma sporadico l’ipotesi è che lo zigote sia normale ma che una stessa linea cellulare subisca due successive mutazioni (teoria dei 2 hits). La teoria è stata convalidata da studi clinici.
La forma ereditaria colpisce bambini molto piccoli. E’ spesso bilaterale e multifocale. La forma sporadica invece si sviluppa in bambini a partire dai 6/7 anni, è monolaterale e monofocale.
· Osservazioni genetiche: se i geni mutati vengono trasmessi in linkage con marcatori (es. microsatelliti) si può studiare la relazione tra malattia e presenza del marcatore associato al gene malato. Analizzando il sangue periferico si nota nelle forme familiari la presenza dell’allele malato. Poi, chiaramente, perché si sviluppi il tumore è necessaria un’ulteriore mutazione.
La penetranza di una malattia indica la percentuale di individui con l’allele mutato che prima o poi sviluppano tumore.
RB
RB è una proteina implicata nel primo check point, quello cioè che blocca la cellula in fase G1. Se la proteina è ipofosforilata il ciclo non va avanti poiché RB forma un complesso con il fattore trascrizionale E2F che viene inattivato.
Se RB è fosforilato dal complesso CDK/ciclica esso si stacca da E2F che può così fungere da fattore trascrizionale. Questo evento consente alla cellula di superare il primo check point.
La proteina RB può non essere presente se sono presenti mutazioni geniche ma può anche essere inattivata se forma complessi con proteine virali, spiegando l’azione cancerogenica di queste proteine virali:
E7 dell’HPV
E1a dell’adenovirus
LT dell’SV40
Anche P53 è inattivata se complessata con proteine virali.
P53
E’ un fattore trascrizionale. Ha un dominio che lega il DNA e media l’attività di regolazione della trascrizione mentre con un altro dominio lega altre proteine per formare omotetrameri.
Quando si verifica un danno non letale al Dna cellulare P53 si attiva e stimola la trascrizione di geni come p21 cip (inibitori delle CDK) che arresta la cellula in fase G1 o GADD45 che ripara il DNA.
Se il danno viene riparato riprende il ciclo, se esso invece è troppo grave allora un’altra possibilità che P53 ha è quella di attivare dei circuiti apoptotici mediando l’espressione di BAX ed inibendo la trascrizione della proteina anti-apoptotica BCL-2.
Quando si verifica un danno al DNA si attivano però due circuiti che fosforilano in due posizioni tipiche P53 rendendola resistente all’ubiquitinazione (proteolisi). Altrimenti l’emivita di P53 sarebbe bassissima. P53 mutata (e inattiva) è più stabile della forma normale e di conseguenza può essere messa in evidenza con metodi immunoistochimici.
MDM2 è una proteina che blocca l’attività di P53 con meccanismo a feedback negativo: la sua espressione è mediata da P53 e il suo effetto è quello di inibire P53 stessa. P19ARP lega MDM2 sottraendola al legame con P53. Se P19 non è funzionante la P53 rimane inattiva e perde la sua funzionalità.
La perdita di funzione di P53 è molto grave perché può originare un clone di cellule con mutazioni successive a danno del DNA che però non vengono riparate.
Quasi il 50% dei tumori sporadici presentano una mutazione che interessa p53.
L’alterazione di p53 in cellule della linea germinale provoca nella prole la sindrome di LI-FRAUMENI caratterizzata da tumori alle ossa e dei tessuti molli in genere.
APC
E’ il gene responsabile della poliposi adenomatosa del colon (FAP). Questa proteina contrae rapporti con la β catenina favorendone la degradazione. In questa maniera la β catenina non può più svolgere il suo ruolo di fattore trascrizionale nei confronti di geni coinvolti nella replicazione cellulare. Ovviamente se APC è inattiva o malfunzionante la replicazione cellulare avverrà più facilmente.
Segni associati alla poliposi del colon sono l’ipertrofia dell’epitelio pavimentose della mandibola ed osteomi multipli della mandibola. Ci sono inoltre alcune varianti della FAP come la sindrome di Torcout nella quale compaiono tumori cerebrali.
L’alterazione dell’APC è un evento precoce tanto che questa alterazione si rivela già allo stato di adenoma. Tuttavia questa alterazione non è di per sé sufficiente allo sviluppo del tumore ma deve essere seguito questo percorso:
Epitelio normale
Alterazione di APC
Displasia a livello dell’epitelio ghiandolare
Demetilazione del DNA (evento epigenetico che riguarda alcuni promotori)
Adenoma precoce
Alterazione di RAS
Adenoma intermedio.
