Nei primi passaggi di purificazione delle proteine è spesso utile impiegare tecniche non cromatografiche di frazionamento selettivo, ad alta capacità. Frazionamento con sali. Si basa sulla precipitazione delle proteine per ‘salting out’. I sali ad alte concentrazioni tendono a diminuire la solubilità proteica attraverso meccanismi piuttosto complessi.
Si sa che gli ioni (anioni e cationi) liberati dalla dissociazione di un sale in acqua interagiscano fortemente con le molecole di solvente, aumentandone la tensione superficiale. Ad alte concentrazioni, gli ioni interagiscono anche con l’acqua d’idratazione delle proteine, ovvero quello strato di molecole di solvente che si trovano a contatto diretto con la superficie proteica e che si considerano abbastanza localizzate ed ordinate (soprattutto a livello di zone idrofobiche della superficie proteica). Si ritiene che gli ioni agiscano da ‘disidratanti’, sfavorendo le interazioni proteina-acqua d’idratazione e favorendo, per converso, le interazioni soprattutto idrofobiche proteina-proteina. Di conseguenza, le proteine finiscono per aggregare e precipitare.
Questo effetto non dipende semplicemente dalla forza ionica, ma dal tipo specifico di sale usato (di solito si usa il solfato d’ammonio, che è particolarmente efficace e solubile fino a circa 4 M) nonché dal tipo di proteina (proprietà elettrostatiche della superficie, capacità di interagire direttamente con gli ioni etc.). Quindi, proteine diverse tenderanno a precipitare a concentrazioni diverse di sale.
Di questo si approfitta per un frazionamento: se, come nell’esempio a lato, è noto che una data proteina precipita a concentrazioni di solfato d’ammonio prossime al 40% di saturazione (1.6 M), si possono precipitare le proteine meno solubili aggiungendo sale all’estratto fino ad un 35% di saturazione (1.4 M) e centrifugando. Al sovranatante si può poi aggiungere ulteriore solfato d’ammonio fino al 45% (circa 1.8 M) precipitando la proteina d’interesse ma non altre proteine più solubili.
Frazionamento con solventi organici. Sfrutta la differente solubilità delle proteine in soluzione miste acqua-solventi organici (quali etanolo od acetone). Il solvente abbassa la costante dielettrica del mezzo, favorendo le interazioni elettrostatiche (anziché idrofobiche) proteina-proteina. L’effetto è in qualche modo opposto a quello descritto sopra, e simile al ‘salting in’ (il fenomeno per cui in acqua distillata o comunque a forza ionica molto bassa la solubilità proteica diminuisce). Il problema con l’uso dei solventi organici è che si ha in genere un certo grado di denaturazione proteica.
Trattamento termico. Ogni proteina ha una differente temperatura di denaturazione. Quando si debbano purificare proteine particolarmente termostabili (ad es., una proteina da organismo termofilo clonata in un batterio mesofilo) è possibile eliminare per denaturazione termica gran parte delle altre proteine riscaldando per 15-20’ ad una temperatura di poco (5-10°C) inferiore alla temperatura di denaturazione della proteina d’interesse. Le proteine denaturate vengono poi eliminate tramite centrifugazione. Anche il pH è una variabile che può essere utilizzata: se la proteina è stabile a pH <3 o >10, si può denaturare buona parte delle altre proteine incubando la miscela a quei pH estremi.
Classificazione in base al meccanismo di separazione:
• Cromatografia di assorbimento,
• Cromatografia di ripartizione,
• Cromatografia a scambio ionico,
• Cromatografia di affinità,
• Cromatografia ad esclusione.
1 Cromatografia di assorbimento:
• si basa sul fatto che alcuni materiali solidi hanno la capacità di trattenere le molecole alla loro superficie (forze di Wander –Waals). I diversi analiti si ripartiscono tra fase mobile e assorbente in modo diverso a seconda delle relative forze di interazione. Può essere condotta sia su strato sottile che su colonna. Tipico assorbente è silice. Il gel di silice viene preparato da silicato di Na+ + HCl con formazione di SiO4 ( tetraossido di silicio).
2 Cromatografia di ripartizione:
• Nella cromatografia di ripartizione sia la FS che la FM sono liquide. La separazione avviene sulla base del coefficiente di ripartizione. La Fase S è legata covalentemente a un supporto solido ( carta, silice ecc.). Nella cromatografia di ripartizione in fase normale, la FS è polarizzata e la FM è relativamente non polare. Vengono eluiti in ordine di polarità crescente.
4 cromatografia a scambio ionico:
• Il principio è l’attrazione tra molecole cariche di segno opposto. Possiamo separare Acidi nucleici, proteine ecc.
La separazione di miscele viene condotta su colonna ,
impaccata di resina,
Resine a scambio cationico ▬▬►il gruppo ionizzabile è il carbossimetile ( ─ ).
Resine a scambio anionico ▬▬►il gruppo ionizzabile è il dietilaminaetano (+ ).
3 Cromatografia di affinità:
• Non si basa sulle differenze nelle proprietà. Viene utilizzata per la separazione di anticorpi.
4 Gas-cromatografia:
• La Fase M e le sostanze da separare sono allo stato gassoso.
La fase S è rappresentata da un solido con proprietà.
assorbenti o un liquido non volatile fissato su un supporto solido inerte.
5 Cromatografia ad esclusione o gel filtrazione:
• La Fase S è rappresentata da una fase liquida intrappolata all’interno di una struttura porosa,
• La Fase M è liquida.
Le sostanze si separano in base alle dimensioni molecolari. I setacci molecolari, reticoli tridimensionali porosi sulla base dei quali si separano i materiali, sono rappresentati da gel d’agarosio, (sephadex), destrano (sephacril),cellulosa, poliacrilammide. Sono usati per la separazione di proteine.
≈≈≈≈≈≈ CALORE quando il gel si rappende
≈≈≈≈≈≈ + ────► ξξξξξξξ ─────────► §§§§§§
≈≈≈≈≈≈ ◄──── ξξξξξξξ §§§§§§
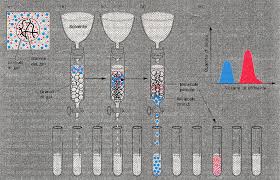
Polimero si formano si è formato un semplice legami crociati reticolo tridimensionale (AGAROSIO) con pori ben precisi. La colonna viene impaccata con gel e solvente. Le particelle di piccole dimensioni entreranno nei pori della matrice e quindi sono ritardati nel loro percorso verso la fine della colonna (comportamento diametralmente opposto al movimento degli acidi nucleici all’elettroforesi su gel d’agarosio).
Quelle che fuoriescono per prima sono invece le particelle di più grosse dimensioni (non entrano nei granuli ) perché attraversano il volume vuoto V0 che è il volume esterno ai granuli.
Vi è il volume incluso nei granuli di gel:
Vt = Vi + V0
Per molecole intermedie : Ve = V0 + Kd +Vi
Soluto grande ════► Kd = 0 Ve = V0,
Soluto piccolo ════► Kd = 1 Ve = V0 + Vi = Vt,
Soluto intermedio ════► 0 ≤ Kd ≤ 1.
Cromatografia ad esclusione : esperienza di laboratorio:
FS : Sephadex G75.
FM : NaCl 50 mM.
Miscela di citocromo c + 2,4 dinitrofenolo.
Cromatografia ad esclusione o gel filtrazione.
esperienza in laboratorio:
FS : sephadex G75,
FM : NaCl 50 mM,
Componenti da separare: Miscela di citocromo c + 2,4 dinitrofenolo
Citocromo c pm 12.000 d,
2,4 dinitrofenolo 184,1 d.
Eluati: Si è scelto l’uso di questi due composti colorati al fine di evidenziare attraverso l’esperienza visiva del colore il principio che è alla base dell’esperienza stessa.
Si raccoglie prima il citocromo c (colore rosso) perché di peso molecolare maggiore (12.000) , poi il 2,4 dinitrofenolo (di colore giallo) perché di peso molecolare minore (184.1) rispetto al citocromo c. Questo perché il 2, 4 dinitrofenolo rimane imbrigliato nel Sephadex. Invece il citocromo c fuoriesce per prima perché attraversa il volume vuoto ( v )che è il volume esterno ai granuli.
Protocollo Purificazione di Proteine:
Spesso l’interesse della ricerca biochimica si concentra sulle singole macromolecole biologiche, in particolare le proteine. Per studiare le proprietà chimiche, fisiche e biologiche di queste molecole è ovviamente necessario procedere dapprima alla loro purificazione, che può essere raggiunta mediante una serie di tecniche.
Tre fattori sono importanti nello sviluppare un protocollo di purificazione:
Purezza desiderata: Il tipo di studi che si intendono eseguire determina il grado di purezza da raggiungere. Ad es., se si vuole ottenere semplicemente una preparazione di enzima per usi industriali il grado di purezza non dovrà essere necessariamente elevato (alcune preparazioni commerciali di enzimi consistono essenzialmente di brodi di fermentazione concentrati ed addizionati di agenti stabilizzanti). Per studi di tipo cinetico o termodinamico, la purezza dovrà essere maggiore, e soprattutto la preparazione proteica deve essere priva di quei materiali (ad es., inibitori enzimatici) che potrebbero interferire con gli esperimenti. Per materiale da destinare a studi strutturali la purezza e l’omogeneità della preparazione dovranno essere massime, vicine al 100%.
Quantità di proteina desiderata: È sempre importante partire da un materiale relativamente ricco della proteina che si intende purificare (per questo, quando possibile in pratica, è particolarmente conveniente purificare proteine over-espresse in microrganismi). La scelta di diverse strategie di purificazione dipende poi in larga parte da quanto di questo materiale si voglia o debba utilizzare. Ad es., per grandi quantità di materiale si dovrà ricorrere a tecniche che consentano di processare volumi elevati (ad es., la precipitazione selettiva o la cromatografia a scambio ionico) ma non tecniche come la cromatografia di affinità che si applica in genere a volumi più ridotti.
Costi della purificazione: Anche il costo dei materiali da utilizzare (quali gli agenti precipitanti, le resine cromatografiche etc.) può essere limitante per preparazioni su larga scala. Ad es., le resine per cromatografia ad affinità sono molto costose, ma consentono un’elevata risoluzione.
Le tecniche di isolamento delle proteine sfruttano le grandi diversità di caratteristiche fisico-chimiche fra queste molecole:
– carica,
– idrofobicità,
– dimensioni,
– affinità per specifici leganti,
– solubilità,
– stabilità al pH ed alla temperatura.
Nel complesso, le caratteristiche uniche di ogni proteina la rendono isolabile da tutte le altre, attraverso una serie di tappe purificative (che possono secondo i casi oscillare tra 2-3 e 9-10). Un buon protocollo di purificazione dovrebbe essere riproducibile e tendere ad ottenere la purezza desiderata attraverso il minor numero possibile di passaggi purificativi.
L’esigenza di ottenere un prodotto puro e l’esigenza di una buona resa finale possono a volte essere in contrasto, poiché in ogni passaggio purificativo si perde una certa quantità della proteina d’interesse. L’ordine nel quale le diverse tecniche vengono applicate è essenziale.
La sequenza delle tappe dovrebbe essere tale che il materiale ottenuto alla fine di un dato passaggio costituisca un buon materiale di partenza per
la tappa successiva: In genere, si tende a far precedere passaggi ad alta capacità/ bassa risoluzione seguiti da passaggi con tecniche a maggior risoluzione (che però di solito implicano minore capacità). In tutti i protocolli, le varie fasi della purificazione sono accompagnate da test per verificare la quantità (nel caso di un enzima, stimata sulla base dell’attività totale) e la purezza (nel caso di un enzima, espressa in termini di attività specifica) della proteina. Spesso, inoltre, è indispensabile ricorrere a tecniche per concentrare la frazione proteica del campione e/o per dializzare il campione stesso. (Dializzare significa rimuovere o sostituire molecole a basso peso molecolare contenute nella soluzione proteica).

